Plus d’un million de personnes en France, et 40 millions dans le monde, sont atteintes par la maladie d’Alzheimer. La première description de cette pathologie par le Docteur Alois Alzheimer date de 1906, il y a plus de 100 ans. À ce jour, aucun traitement curatif n’est encore disponible. Alors, qu’avons-nous appris et qu’ignorons-nous encore sur cette maladie ? Quels sont les enjeux de la recherche ?
Définition et diagnostic de la maladie d’Alzheimer
La maladie d’Alzheimer est une maladie neurodégénérative, c’est-à-dire caractérisée par la mort de neurones. Elle se manifeste cliniquement par des troubles neurocognitifs1 progressifs et représente la forme de démence la plus commune (60 à 80 % des cas). Les symptômes débutent le plus souvent par des oublis ponctuels puis évoluent vers des difficultés dans les tâches routinières, des troubles du sommeil, des changements d’humeur, des perturbations du langage et des pertes de mémoire importantes. En particulier, la mémoire de travail et la mémoire sémantique sont fortement dégradées 2, de même que la mémoire épisodique 3. Il s’agit d’une maladie dévastatrice, qui touche près d’une personne sur cinq de plus de 75 ans en France (environ 1 million de cas au total), et 40 millions de personnes dans le monde. Le nombre de malades est en forte augmentation (Figure 1), et selon l’Organisation mondiale de la santé, la prise en charge de la maladie d’Alzheimer devrait coûter plus de 2 000 milliards de dollars américains en 2030 si aucun traitement efficace n’est proposé d’ici là 4.
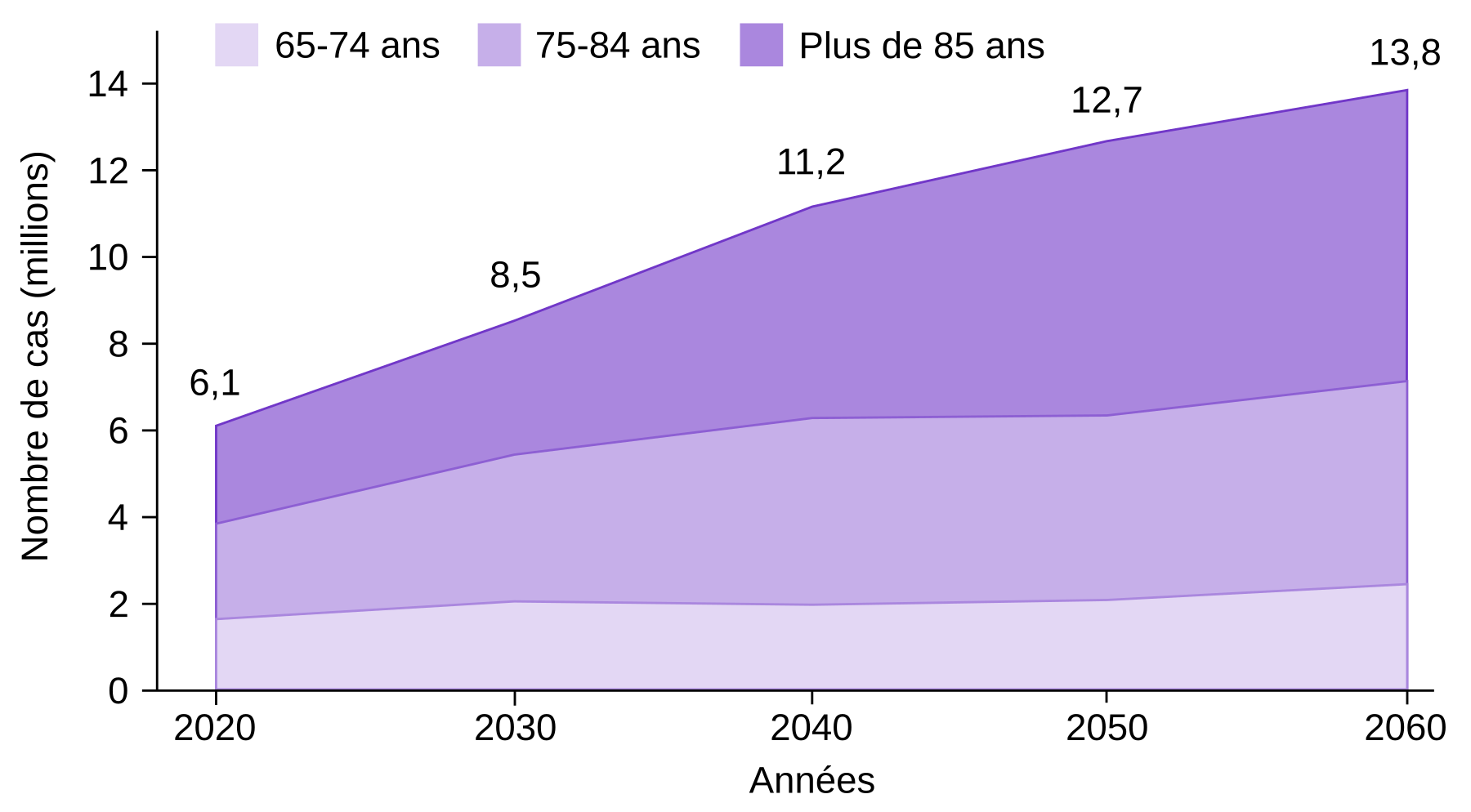
Source des données : Rajan et coll., 2021, Alzheimer's & Dementia.
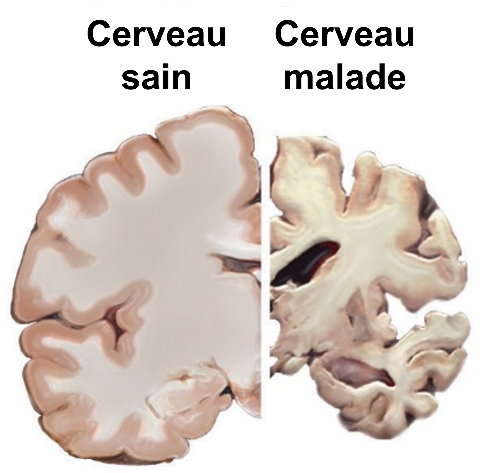
En France, le diagnostic de la maladie d’Alzheimer repose sur des tests cognitifs et neuropsychologiques évaluant les fonctions cérébrales touchées, tandis que des techniques d’imagerie cérébrale telles que l’IRM permettent de caractériser la diminution du volume cérébral (également visible post-mortem, voir Figure 2) et d’écarter d’autres causes possibles de troubles cognitifs (tumeurs, pathologies vasculaires…). Dans certains cas (sujets jeunes ou formes atypiques), des prélèvements du liquide céphalo-rachidien peuvent être réalisés, pour doser des protéines associées à la maladie comme Tau ou Aβ.
Deux marqueurs associés à la maladie d’Alzheimer : le peptide β-amyloïde et la protéine tau phosphorylée
La maladie d’Alzheimer se caractérise notamment par l’accumulation de deux protéines dans le cerveau : le peptide β-amyloïde (Aβ), sous forme de plaques extracellulaires et la protéine tau phosphorylée (p-tau), sous forme de neurofibrilles intraneuronales (Figure 3).
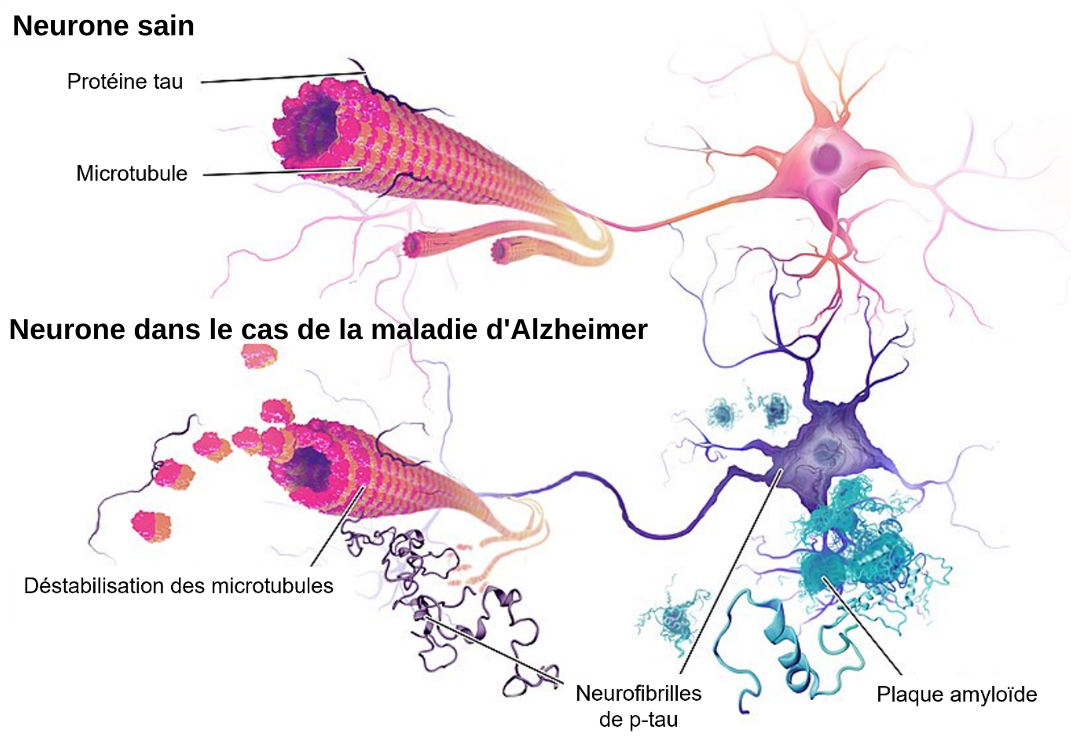
Le peptide Aβ est formé à partir de la protéine APP (Aβ protein precursor, codée par le gène APP sur le chromosome 21). De nombreux types de peptides Aβ existent. Ils sont produits par clivage séquentiel de l’APP, par les enzymes β-sécrétase, puis γ-sécrétase. Le peptide Aβ40 (40 acides aminés) est majoritairement produit, tandis que le peptide Aβ42 (42 acides aminés) constitue seulement environ 10 % du total d’Aβ mais est retrouvé en proportion importante dans les plaques amyloïdes. Le peptide Aβ42 s’agrège dans le milieu cérébral en oligomères solubles, puis en fibrilles et enfin sous forme de plaques extracellulaires, les plaques amyloïdes 1. Ces plaques ont d’ailleurs été décrites, dès 1906, par Alois Alzheimer. La fonction physiologique des peptides Aβ solubles n’est pas totalement comprise, mais ils semblent avoir des propriétés anti-microbiennes 2 et être impliqués dans l’activité synaptique 3. Cependant, les oligomères amyloïdes sont toxiques pour les neurones. Les connaissances sur les propriétés structurelles et neurotoxiques des formes solubles d’Aβ restent à approfondir.
La protéine Tau constitue le second marqueur pathologique de la maladie d’Alzheimer. Codée par le gène MAPT (chromosome 17), la protéine Tau est principalement exprimée dans les neurones et appartient à la famille des protéines associées aux microtubules (MAP). Sa fonction principale est de favoriser l’assemblage et la stabilité des microtubules des axones. Des modifications post-traductionnelles, notamment la phosphorylation de plusieurs sites régulés par différentes kinases, contribuent à moduler la structure de la protéine Tau, et ainsi sa fonction et sa localisation. Dans la maladie d’Alzheimer, des modifications post-traductionnelles de Tau, dont l’hyperphosphorylation, conduisent à son détachement du microtubule puis à son assemblage en filaments et finalement en neurofibrilles dans les neurones. Ce processus perturbe le transport axonal, contribuant ainsi à la perte synaptique et à la neurodégénérescence. Les déficits cognitifs corrèlent mieux avec la charge en neurofibrilles qu’avec celle en plaques amyloïdes 4. L’accumulation anormale de ces protéines est également associée à d’autres maladies neurodégénératives, regroupées sous le terme de tauopathies : les démences fronto-temporales, la maladie de Pick, la paralysie supranucléaire progressive…
Formes familiales et formes sporadiques de la maladie d’Alzheimer
La maladie d’Alzheimer peut se présenter sous une forme familiale ou sous une forme dite sporadique. La première, rare (<1 % des cas), est autosomique dominante et causée par des mutations monogéniques dans les gènes clés du métabolisme de l’Aβ : APP, PSEN1 ou PSEN2 (présénilines 1 et 2, qui composent l’enzyme γ-sécrétase). Ces mutations sont suffisantes au développement de la maladie d’Alzheimer familiale 5. Cette forme se manifeste souvent précocement (avant 65 ans, parfois dès 40 ans) et présente une évolution clinique rapide.
Les formes sporadiques, très majoritaires, ne sont pas des maladies génétiques à proprement parler puisqu’elles ne se transmettent pas de manière systématique de génération en génération. En revanche, certains variants génétiques constituent des facteurs de risque de développer la maladie d’Alzheimer. Par exemple, les porteurs homozygotes de l’allèle APOE4 du gène APOE présentent un risque plus de dix fois plus élevé de développer la maladie par rapport aux porteurs homozygotes de l’allèle APOE3 (le plus fréquent). L’allèle APOE2, lui, est associé à une diminution du risque 6. Grâce au développement des outils de séquençage depuis les années 2000, des études d’association pangénomique (GWAS en anglais, pour Genome-Wide Association Studies) ont identifié plus de 70 loci génétiques associés à la maladie d’Alzheimer 7. Ces gènes sont principalement impliqués dans le métabolisme des lipides et les mécanismes d’endocytose. Cependant, les mécanismes biologiques expliquant l’impact des variants dans ces 70 loci restent très largement inconnus et constituent un enjeu majeur de la recherche.

L’hypothèse amyloïde
Formulée en 1992, l’hypothèse amyloïde considère que l’accumulation de peptides Aβ est l’évènement central dans le développement la maladie d’Alzheimer. Selon cette hypothèse, l’accumulation de peptides amyloïdes jusqu’à la formation de plaques amyloïdes (favorisée par l’âge, des facteurs de risques génétiques ou environnementaux) mènerait à la phosphorylation anormale de la protéine Tau, à l’origine de la dégénérescence 1. Cet enchaînement est aussi appelé « cascade amyloïde » (Figure 4).
L’hypothèse amyloïde a été formulée suite à la découverte des mutations liées à la forme familiale de la maladie, toutes situées dans des gènes impliqués dans le métabolisme de l’Aβ. Un autre argument en faveur de l’hypothèse amyloïde est le cas des personnes atteintes de trisomie 21, présentant trois copies du chromosome 21, et donc trois copies du gène APP. Ces individus présentent pour la plupart des plaques amyloïdes et les neurofibrilles de p-tau dès 40 ans, et la maladie d’Alzheimer touche près de 90 % des plus de 65 ans 2. La présence d’une copie additionnelle du gène APP, directement liée à l’augmentation du risque de développer la maladie d’Alzheimer, est cohérente avec l’hypothèse amyloïde.
Comme la protéine Tau seule (en l’absence d’Aβ) est associée à la neurodégénérescence dans d’autres démences, il a été avancé que l’accumulation d’Aβ induit la pathologie tau, ce qui conduirait à la mort neuronale et expliquerait les symptômes cliniques.

Dans le cadre de l’hypothèse amyloïde, de nombreux essais cliniques ont cherché à moduler la voie Aβ (inhibition des acteurs de la production d’Aβ, immunothérapie anti-Aβ). Les résultats ont cependant été mitigés, et le lien entre Aβ et cognition reste mal compris 1. Récemment, des anticorps anti-Aβ tels que le donanemab (Eli Lilly), l’aducanumab (Biogen), et le lecanemab (Biogen et Eisai) ont été approuvés ou sont en cours d’approbation aux États-Unis par l’Agence américaine des produits alimentaires et médicamenteux (Food and Drug Administration). L’aducanumab et le lecanemab visent les formes oligomériques préfibrillaires tandis que le donanemab cible les plaques. Ces traitements semblent avoir un effet léger, mais significatif, sur le déclin cognitif des patients. Ils présentent cependant des effets secondaires importants (par exemple le taux d’incidence d’œdème cérébral varie entre 10 et 35 % selon la molécule 2). Par ailleurs, leur coût est extrêmement élevé (26 000 dollars par an 3). L’Agence européenne des médicaments s’était d’abord prononcée contre la mise sur le marché du lecanemab en juillet 2024, jugeant les risques trop importants par rapport à la faible amélioration cognitive apportée par le traitement 4. Cependant, une réévaluation des études a permis de montrer que ces risques étaient surtout élevés chez les patients homozygotes pour l’allèle ApoE4. Pour les patients ne possédant qu’une ou aucune copie de l’allèle ApoE4, les bénéfices sont jugés supérieurs aux risques, ce qui a conduit, en novembre 2024, l’Agence européenne des médicaments à se prononcer en faveur de la mise sur le marché du lecanemab 5.
La réduction de la charge amyloïde ne semblant pas entraîner d’amélioration radicale des déficits cognitifs des patients, il est important de réévaluer l’hypothèse amyloïde, qui a absorbé d’importants moyens de recherche au cours des trente dernières années.
D’autres pistes pour expliquer la maladie d’Alzheimer
Depuis sa formulation au début des années 90, l’exploration de l’hypothèse amyloïde, passant par la mise au point de médicaments ciblant les plaques amyloïdes n’a, à ce jour, pas permis de révolutionner le traitement de la maladie d’Alzheimer. Au moins deux raisons pourraient expliquer cet échec relatif :
- Il est probable que les plaques amyloïdes ne constituent qu’un marqueur tardif de la maladie et que des événements antérieurs soient à l’origine du développement de la pathologie.
- En plus des plaques amyloïdes, il est nécessaire de cibler d’autres mécanismes physiopathologiques pour freiner le développement de la maladie.
Neuro-inflammation et implication des cellules gliales
Alors que la recherche s’est longtemps concentrée sur les neurones, il devient évident que les cellules gliales (microglies, astrocytes et oligodendrocytes)6 jouent aussi un rôle crucial dans la maladie d’Alzheimer. Les microglies et astrocytes sont des cellules essentielles pour l’homéostasie cérébrale, elles sont par exemple impliquées dans le métabolisme et l’élimination des déchets du cerveau. Dans les stades tardifs de la maladie d’Alzheimer, l’inflammation chronique du tissu cérébral causée par les lésions entraîne une activation des microglies et astrocytes, modifiant leur phénotype, leur morphologie et leurs fonctions. En particulier, la sécrétion de cytokines pro-inflammatoires augmente, aboutissant à un contexte de neuro-inflammation. L’étude du transcriptome des cellules gliales a montré d’importantes modifications de celui-ci dans la maladie d’Alzheimer, à la fois dans des modèles animaux 78 et dans des cerveaux post-mortem 9. De plus, de nombreux gènes associés à la maladie d’Alzheimer (APOE, TREM2, SPI1, BIN1) sont principalement exprimés par les microglies et/ou les astrocytes 10, soulignant l’importance des cellules gliales dans la maladie.
Le rôle précis de ces cellules dans la maladie est cependant très complexe : certaines populations de cellules gliales semblent avoir un effet protecteur tandis que d’autres seraient néfastes, selon la progression de la pathologie. Par exemple, les microglies exprimant TREM2 sont impliquées dans l’élimination des plaques amyloïdes 11, mais l’inhibition de TREM2 atténue la pathologie Tau et la neurodégénérescence, suggérant alors un effet globalement délétère de ce gène 12. Une hypothèse est que ces microglies sont protectrices dans les premiers stades de la pathologie, mais changent progressivement de fonction avec leur activation chronique 13. De même, le rôle des astrocytes activés, dits « réactifs », reste largement méconnu, bien que des travaux suggèrent leur participation à l’internalisation et la dégradation du peptide Aβ, et à la propagation intercellulaire de la protéine Tau 14. Par ailleurs, des travaux montrent des échanges bidirectionnels de protéine Tau entre neurones et astrocytes dans un modèle murin 15. En effet, il ne faut pas oublier que les différents types cellulaires du cerveau interagissent entre eux de manière complexe et permanente. Les astrocytes sont essentiels à l'activité des neurones, par exemple en régulant les synapses et en assurant l’approvisionnement des neurones en cholestérol. L’étude de leur implication est donc très pertinente dans la maladie d’Alzheimer, pour laquelle une perturbation du métabolisme des lipides est un risque génétique. Microglies et astrocytes dialoguent également : des études suggèrent que les microglies activées induisent la réactivité des astrocytes 16, et vice-versa 17, ce qui contribuerait à l’aggravation de la pathologie.
L’implication des cellules gliales dans la maladie d’Alzheimer ainsi que dans les autres maladies neurodégénératives et le vieillissement reste à préciser. De manière intéressante, l’activation de la voie JAK/STAT dans les astrocytes réactifs semble néfaste dans la maladie d’Alzheimer 18, mais protectrice dans la maladie de Huntington (une autre maladie neurodégénérative) 19. Cela souligne la complexité des mécanismes, qui dépendent du contexte pathologique.
Pathologie vasculaire et maladie d’Alzheimer
La barrière hémato-encéphalique constitue une interface entre le cerveau et le sang. Elle protège en particulier le cerveau de composés potentiellement nocifs provenant du sang. Cette barrière est constituée de cellules endothéliales, de cellules musculaires lisses (péricytes au niveau des capillaires) et d’astrocytes. Sa fonction de filtre est assurée par les jonctions serrées et adhérentes qui unissent les cellules endothéliales. Des transporteurs spécifiques (GLUT1 pour le glucose par exemple) assurent le passage de nutriments du sang vers le cerveau 20. D’autres transporteurs exportent des composés potentiellement toxiques et contribuent ainsi à leur élimination. Par exemple le transporteur P-gp permet l’élimination du peptide Aβ 21.
Dans la maladie d’Alzheimer, ainsi que dans d’autres pathologies du cerveau, des études d’imagerie cérébrales montrent une augmentation de la perméabilité de la barrière hémato-encéphalique, causée par la détérioration des cellules endothéliales, des péricytes et des astrocytes, ce qui provoque des microhémorragies et l’infiltration de cellules immunitaires 22. De manière intéressante, ces perturbations de la barrière hémato-encéphalique apparaissent souvent lorsque les troubles cognitifs sont encore légers et pourraient ainsi constituer un biomarqueur précoce de la maladie 23. Au niveau cellulaire, la maladie d’Alzheimer se caractérise par une perturbation de l’expression des transporteurs des cellules endothéliales, avec une baisse d’expression des transporteurs d’export et une hausse de ceux d’import du peptide Aβ 24. Les autres barrières cerveau-périphérie comme les plexus choroïdes ou les méninges sont également importantes dans ces mécanismes d’élimination.
De plus, de nombreux patients atteints de la maladie d’Alzheimer présentent également une angiopathie amyloïde cérébrale, c’est-à-dire l’accumulation de dépôts amyloïdes dans les vaisseaux cérébraux 25. L’étude de la barrière hémato-encéphalique dans la maladie d’Alzheimer a donc des enjeux importants, d’autant plus que les œdèmes et microhémorragies au niveau des vaisseaux cérébraux constituent les risques principaux des immunothérapies anti-amyloïdes comme le lecanemab. De plus, ces effets secondaires affectant préférentiellement les homozygotes APOE4, il est nécessaire de mieux caractériser l’interaction d’APOE avec la barrière hémato-encéphalique
Implication de l’immunité périphérique : exemple du rôle des lymphocytes T
Alors que le cerveau a longtemps été considéré comme bénéficiant d’un privilège immunitaire, c’est-à-dire isolé des réponses du système immunitaire du reste du corps, il est admis aujourd’hui que des cellules immunitaires dialoguent activement avec le cerveau au niveau de niches immunitaires situées à l’interface entre cerveau et périphérie 26. Dans la maladie d’Alzheimer, des travaux récents ont démontré une infiltration de lymphocytes T dans le cerveau des patients malades. Alors que la présence de lymphocytes T CD8+ clonaux dans le liquide céphalo-rachidien de patients avait été décrite 27, une étude utilisant des cellules humaines dérivées de cellules souches dans une puce microfluidique in vitro a montré que les lymphocytes CD8+ infiltrent le compartiment cérébral et y interagissent avec les microglies, ce qui accélère la neurodégénérescence 28. L’inhibition de cette interaction permet de limiter la mort neuronale. Des résultats similaires ont été obtenus dans un modèle murin de pathologie Tau de la maladie d’Alzheimer 29. Mieux comprendre la communication entre les cellules immunitaires périphériques et le cerveau constitue donc une potentielle piste thérapeutique.
D’autres perturbations corrélées à la maladie d’Alzheimer
En plus des exemples développés ci-dessus, de nombreux autres mécanismes sont associés à la maladie (Figure 5).
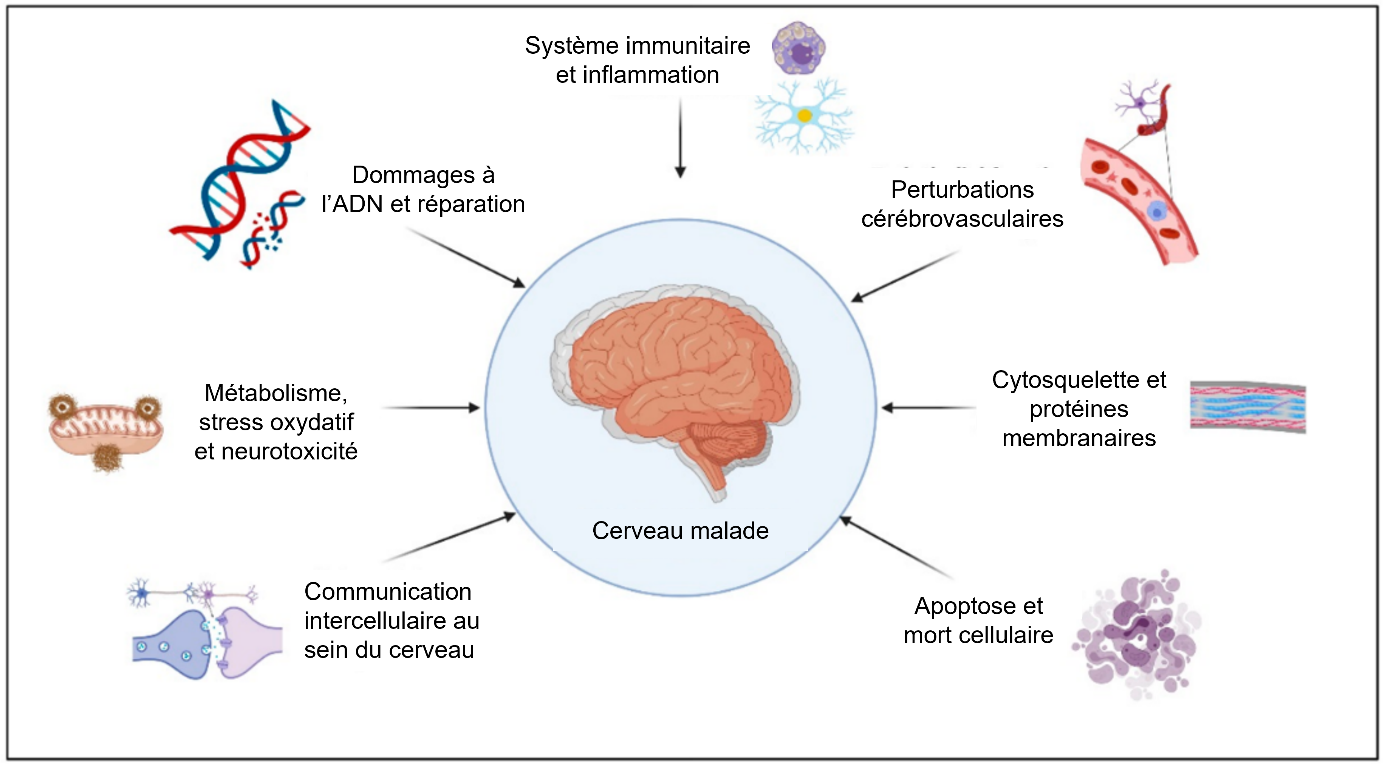
Les rôles des anomalies de l’endocytose et du trafic intracellulaire, du métabolisme des lipides, du microbiote, et des infections sont également explorés. Par exemple, une étude de 2017 a montré une réduction de la biodiversité du microbiote intestinal des patients souffrant de la maladie d’Alzheimer, avec notamment une augmentation de l’abondance des Bacteroidota (anciennement Bacteroidetes) et une diminution de l’abondance des Bacillota (anciennement Firmicutes) 1. Si l’observation d’une telle corrélation ne suffit pas à démontrer la causalité, certains auteurs suggèrent qu’un état de dysbiose pourrait être à l’origine de la neuro-inflammation responsable de la maladie d’Alzheimer 2. De manière plus générale, différents micro-organismes pourraient causer un état d’inflammation du tissu cérébral auquel les cellules répondraient par la production de peptide β-amyloïde, qui présente des propriétés antimicrobiennes. Selon cette conception, la production de peptide β-amyloïde ne serait donc que la conséquence de la réponse à des infections de micro-organismes 3.
Enjeux majeurs : prise en compte de la diversité et de l’environnement
Si les progrès dans la compréhension de la maladie suscitent évidemment de l’enthousiasme, il est essentiel de noter que les études génétiques et cliniques menées se sont concentrées sur des malades caucasiens. Or, du fait de la diversité génétique existant au sein de l’espèce humaine, les mécanismes de la maladie varient. Par exemple, l’allèle APOE4, qui augmente le risque dans les populations caucasiennes, semble avoir un impact moindre dans les populations afro-américaines et hispaniques, mais un effet exacerbé dans les populations japonaises 1. Étudier les mécanismes de la maladie d’Alzheimer dans des populations diversifiées permettra de mieux cibler et traiter les malades.
Il est également important de remarquer que 70 % des patients atteints de la maladie d’Alzheimer sont des femmes. L’espérance de vie plus longue des femmes influence cette différence, mais d’autres facteurs comme la génétique, le système hormonal ou le mode de vie pourraient également y contribuer. De plus, les symptômes cliniques et la progression de la pathologie semblent différer entre hommes et femmes. Par exemple, le déclin cognitif chez les femmes semble plus rapide que chez les hommes 2 ; une autre étude chez 1100 patients suggère que les femmes seraient plus atteintes par les symptômes dépressifs et les troubles du comportement que les hommes 3. Ces connaissances parcellaires sur les différences entre sexes témoignent de l’enjeu à prendre en compte cette variable dans la recherche sur le cerveau et de son vieillissement.
Enfin, un enjeu essentiel de la recherche sur la maladie d’Alzheimer est la prise en compte des facteurs environnementaux. De nombreuses études suggèrent une association entre le risque de développer la maladie et le mode de vie (activité sportive, alimentation, niveau d’éducation… 4), mais également la pollution 5 (Figure 6). Ces études doivent cependant être approfondies pour permettre d’améliorer la prévention de la maladie, l’obstacle principal étant le manque de données. Le développement de différentes biobanques contenant des informations sur le mode de vie permettrait de mieux comprendre la complexité de la maladie d’Alzheimer.

Conclusion
La maladie d’Alzheimer est une maladie complexe, seulement partiellement comprise malgré plus d’un siècle de recherche. Face aux prévisions inquiétantes de l’augmentation du nombre de cas dans les décennies à venir, il est essentiel de poursuivre les efforts de recherche et de développer des modèles biologiques pertinents pour en étudier les mécanismes. Dans une optique de prévention et de prise en charge personnalisée, il est également crucial de s’intéresser à l’impact du sexe, de l’origine géographique et des facteurs environnementaux sur le développement de la maladie.