Les études de cohortes permettent d'identifier les gènes responsables de maladies. Elles furent d'abord limitées, pour des raisons technologiques, à l'identification de maladies monogéniques. Cependant, les progrès réalisés dans la domaine du séquençage ont étendu le champ de ces études à la caractérisation de maladies dépendant de plusieurs gènes mais également de facteurs environnementaux.
On estime aujourd’hui qu’il existe entre 6000 et 8000 maladies humaines d’origine génétique, en lien avec une anomalie dans l’un des 20 000 gènes qui composent le génome humain. Fort heureusement, la quasi-totalité de ces maladies sont rares (fréquence < 1/2000 individus). Souvent il n’existe que quelques dizaines de cas décrits à travers le monde. Par contre si on les considère collectivement, ces maladies rares sont en fait fréquentes et touchent 1 personne sur 20 et, dans 80 % des cas, commencent dès l’enfance.
L’identification des anomalies génétiques à l’origine de ces maladies est donc un enjeu de santé publique et un véritable défi scientifique, mais un prérequis obligatoire pour la compréhension de ces pathologies et l’espoir d’un traitement.
Dans 80 % des maladies génétiques, l’explication réside dans l’altération de la séquence codante d’un gène précis (mutation exonique dans le cas d’une maladie monogénique). Mais il n’est pas rare d’avoir à envisager d’autres mécanismes plus délicats à mettre en évidence : altération de l’expression d’un gène par anomalie de méthylation (défaut d’apposition d’empreinte parentale, disomie uniparentale), par anomalie des séquences promotrices et/ou régulatrices, ou encore par mutation intronique modifiant l’épissage.
Plusieurs approches sont envisageables pour identifier ces gènes, mais l’étude de cohorte est de loin celle ayant eu, et ayant encore, le plus de succès depuis l’avènement de la génétique moléculaire dans les années 90. Le principe est simple, il s’agit à chaque fois, d’identifier dans une cohorte de patients porteurs de la même maladie, les similitudes génétiques partagées par tous les patients malades et absentes chez les personnes saines, permettant ainsi d’en déduire le gène responsable de la maladie.
Les études de liaison ont rencontré de grands succès dans les années 90, où elles ont permis d’identifier les locus impliqués dans de nombreuses maladies monogéniques. Cependant ces études se heurtent aux problèmes des maladies multifactorielles (ayant à la fois une composante génétique et environnementale), plurigéniques (mettant en jeu plusieurs gènes) et à celles causées par des gènes de prédisposition dont la pénétrance et l’expressivité est très variable (tous les individus porteurs de la mutation n’expriment pas le phénotype). Les études actuelles sont également confrontées à des affections toujours plus rares dans des populations étudiées toujours plus grandes avec l’émergence des problèmes de polymorphismes (variant présent dans une population sans caractère délétère).
Études de liaison
Approche historique (années 90)
Au début des années 90, le développement de la biologie moléculaire, avec les techniques de transfert d’ADN (Southern blot), puis l’arrivée du séquençage Sanger, a permis de compléter efficacement la cytogénétique et l’étude strictement chromosomique (caryotype) par l’étude ciblée de la séquence d’ADN pour expliquer les maladies humaines. Ces premières techniques ne permettant d’étudier qu’une infime partie du génome à la fois, il était nécessaire de les compléter par une stratégie permettant de restreindre la « zone de recherche » dans la quête des gènes pathogènes. Si le caractère génétique de ces maladies était clairement établi par l’hérédité récurrente et bien connue, il est apparu évident de développer l’étude de cohortes ou de familles, plutôt que de patients uniques.
L’étude de liaison, ou clonage positionnel, part donc de l’hypothèse, que dans une famille où plusieurs personnes sont atteintes d’une même maladie probablement génétique et dont le mode de transmission est compatible avec une hérédité mendélienne (monogénique), les personnes atteintes doivent partager un facteur génétique familial commun, qui est absent chez les personnes en bonne santé de la même famille. Le principe est donc, par l’analyse de nombreux membres de la famille, d’identifier la région du génome partagée uniquement par les patients malades. Cette région doit contenir le gène responsable de la maladie. Bien sûr, plus le nombre de patients étudiés est important, plus la région retenue est précise et de petite taille, permettant ainsi de se rapprocher de l’identification d’un gène unique. Dans certains cas plusieurs gènes peuvent être compris dans l’intervalle, il faut donc passer par des études fonctionnelles par exemple pour incriminer formellement un seul de ces gènes. Très souvent ces études requièrent quelques dizaines de patients, sur plusieurs familles.
Le principe de la localisation est basé sur :
- la présence de régions répétées fortement polymorphiques (microsatellites), réparties tout le long du génome et possédant un nombre de répétition variable d’un individu à l’autre. Ces régions sont la base des études d’empreinte génétique.
- le brassage génétique par enjambement (crossing-over).
La technique de Southern blot permet dans un premier temps de cartographier les microsatellites, puis le séquençage Sanger sert à identifier les séquences présentes dans la région retenue et à mettre en évidence le gène et la mutation familiale.
De cette manière ont été identifiés de nombreuses mutations à l’origine de maladies monogéniques, parmi les plus fréquentes : amyotrophie spinale, maladie de Huntington, etc.
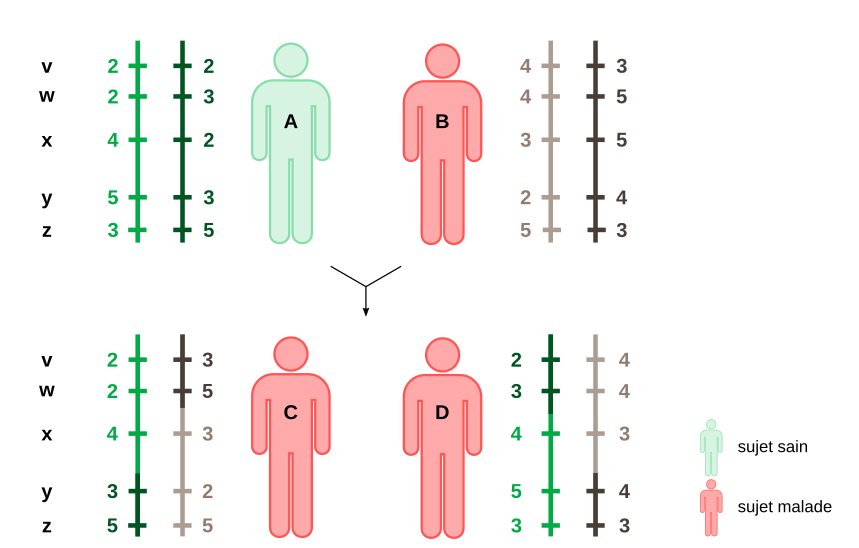
Considérons deux individus A et B, ayant cinq loci microsatellites notés v, w, x, y et z sur le chromosome 10. Pour chaque microsatellite, plusieurs allèles existent en fonction du nombre de répétitions de la séquence. Dans cet exemple fictif, on considère que le nombre de répétitions peut être égal à 2, 3, 4 ou 5. L’individu A est de génotype (2//2 ; 2//3 ; 4//2 ; 5//3 ; 3//5) et l’individu B de génotype (4//3 ; 4//5 ; 3//5 ; 2//4 ; 5//3). Ensemble ils ont deux enfants, C (2//3 ; 2//5 ; 4//3 ; 3//2 ; 5//5) et D (2//4 ; 3//4; 4//3 ; 5//4 ; 3//3). A étant le père et B la mère, le chromosome 10 paternel de C est issu d’un crossing-over entre les loci x et y, tandis que pour son chromosome maternel, il y a eu un crossing-over entre w et x, etc. Comme B, C et D sont malades et que la seule région du chromosome 10 en commun est située après le locus w mais avant le locus z, le gène responsable de la pathologie est situé quelque part sur cet intervalle (donc à proximité du locus x). En pratique, plus le nombre de locus microsatellites est élevé, et plus la localisation du gène est précise.
Gènes et mutations
En recherche, lorsqu’un gène est fortement suspecté d’être à l’origine d’une pathologie (par exemple à l’issue d’une étude de liaison), la mise en évidence de mutations chez les personnes atteintes permet d’incriminer formellement le gène comme étant à l’origine de la pathologie. L’identification des mutations est également nécessaire pour poser le diagnostic chez le patient et proposer un dépistage familial voire prénatal. Si la mutation est bien in fine l’anomalie à l’origine de la maladie, la connaissance précise de celle-ci (délétion, addition, substitution…) ne présente en réalité que peu d’intérêt pratique. En effet il s’agit la plupart du temps de mutations privées, c’est-à-dire que pour une même pathologie et un même gène touché, chaque famille est porteuse de sa propre mutation, qui est différente de celles des autres familles atteintes.
Cependant, il existe de rares cas de mutations récurrentes (tous les patients sont porteurs de la même mutation), comme celles à l’origine de la drépanocytose ou de l’achondroplasie par exemple. Dans ce cas, l’identification de cette mutation permet de repérer une zone de la protéine particulièrement importante pour son fonctionnement.
De manière générale, c’est surtout l’identification du gène concerné plutôt que de la mutation qui est importante pour comprendre les mécanismes à l’origine de la maladie et donc envisager un traitement.
Approche moderne
L’étude de liaison classique telle que décrite ci-avant fonctionne très bien lorsque la maladie est bien caractérisée, récidive dans la famille et est suffisamment fréquente pour constituer des cohortes de patients. De nos jours, les maladies génétiques dont l’étiologie, c’est-à-dire la cause, n’est pas connue sont en général les maladies les plus rares (quelques patients à travers le monde), parfois sans récidive familiale (formes de novo, nécessitant donc d’avoir accès à plus de patients issus de familles différentes), et les moins bien définies (possible confusions entre une maladie unique / un groupe de maladies semblables / des maladies à l’expressivité très variable). Pour ces maladies plus difficiles à cerner, comme pour les maladies plus classiques, l’approche actuelle utilise les outils récents de biologie moléculaire. À savoir, les techniques de séquençage à haut débit (NGS, next-generation sequencing) d’exome ou de génome.
Le principe, moins subtil que le clonage positionnel, est de séquencer dans une cohorte de quelques patients atteints, l’ensemble du génome, ou à défaut de l’exome (ensemble des exons, ce qui représente environ 2% du génome). Il s’agit donc d’une technique générant une quantité très importante de données (voir encadré ci-dessous). L’ensemble des variants de séquence obtenus pour chaque patient (plusieurs milliers) est trié, en éliminant les variants ethniques, et polymorphismes rares déjà décrits dans les bases de données génomiques internationales chez des patients sains. Puis les variants correspondant au mode de transmission attendu sont retenus, et classés selon leur probable effet pathogène (type de mutation, effet délétère prédit in silico). À ce stade plusieurs dizaines de gènes candidats sont retenus et classés également selon leur vraisemblance en tenant compte du rôle physiologique connu des gènes en question. Les informations sont recoupées entre patients atteints afin d’isoler un ou quelques gènes fortement candidats. Les études familiales chez les apparentés sains ne retrouvant pas ces variants confortent l’incrimination de ces gènes. Des études fonctionnelles sont ensuite réalisées (modèle animal transgénique ou KO présentant un phénotype similaire, études biochimiques, cellulaires, transcriptionnelles…).
Données bioinformatiques issues du séquençage à haut débit d’exome ou de génome
La quantité de données générée par le séquençage d’un génome humain est colossale. Puisque chaque base nucléotidique peut prendre la valeur A, T, C ou G et sachant qu’un octet est un ensemble de 8 bits, où chaque bit peut avoir 2 valeurs (1 ou 0), une base est donc codée au minimum sur 2 bits (00, 11, 10 et 01), une paire de bases sur 4 bits. Un octet permet donc d’encoder 2 paires de bases (4 nucléotides). Le génome humain étant porté de manière diploïde, 1 octet ne permet finalement que l’encodage d’une seule coordonnée génomique (4 bases, réparties en 2 couples sens et anti-sens, sur les 2 allèles). Le génome humain étant composé d’environ 3,5 Gpb, chaque individu est donc porteur d’une information de 7 Gpb (diploïdie). L’espace minimal théorique requis pour le stockage est donc de 3,5 Go. Toutefois, le NGS étant sujet à l’apparition d’erreurs aléatoires lors du séquençage, chaque région est séquencée un grand nombre de fois, afin de repérer et corriger ces erreurs. Ainsi il est courant de séquencer simultanément entre 20 et 40 fois chaque région lors d’une analyse de génome (on parle alors d’une profondeur de lecture de 40x). Cela génère bien évidemment 20 à 40 fois plus de données, l’espace nécessaire pour stocker un génome dépasse alors les 100 Go. En pratique ce volume est encore plus important car pour chaque position, ce n’est pas seulement la nature de la base (A, T, G ou C) qui est enregistrée, mais également des données quantitatives (intensité de fluorescence…). Les informations issues des 20 à 40 séquençages du génome sont ensuite recoupées pour constituer un fichier donnant la séquence du génome entier. Pour les analyses cliniques, des fichiers plus légers (quelques Mo), contenant uniquement des informations sur les SNP sont produits.
En pratique, le stockage de To de données, et surtout leur analyse requièrent une grande puissance informatique et l’utilisation de serveurs dédiés, universitaires ou hospitaliers. Parfois des solutions de stockage et d’analyses peuvent être utilisées sur des serveurs externes (cloud) avec les problèmes de confidentialité et de sécurité que cela engendre. Le bioinformaticien est devenu une personne essentielle aux laboratoires utilisant le NGS.
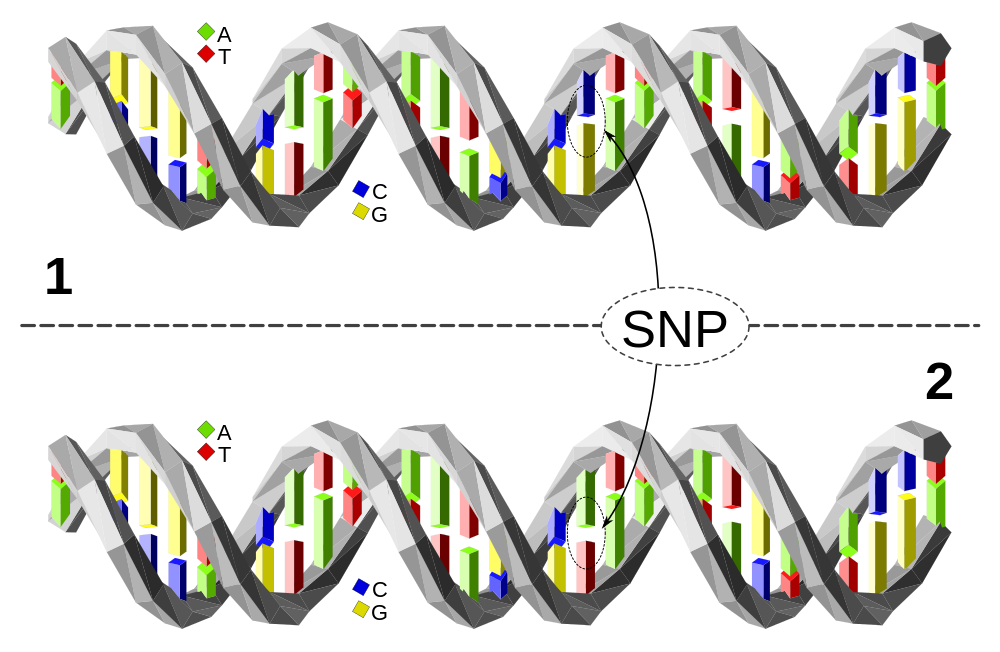
Le long de cette courte séquence, l’ADN des individus 1 et 2 est identique à une paire de nucléotides près : l’individu 1 possède le couple C-G tandis que l’individu 2 présente le couple A-T. Cette variation d’une paire de nucléotides, si elle est présente chez au moins 1 % de la population, est appelée polymorphisme mononucléotidique (SNP en anglais, à prononcer « snip »). En fonction de l’endroit où se situe la variation elle peut avoir un effet sur le phénotype ou non.
SNP et puces SNP
À l’interface entre l’approche NGS, et l’approche GWAS (détaillée ci-après) se trouvent les puces SNP. Version moderne de l’étude de microsatellites, les SNP (single nucleotide polymorphism) sont des régions variables du génome (à l’image des microsatellites), où la variation ne va concerner qu’une seule base azotée (Figure 2). Il s’agit d’un polymorphisme mononucléotidique, ou SNP. Des millions de SNP sont connus sur le génome humain. La combinaison des SNP portés par un individu peut être utilisée comme une empreinte génétique pour l’identifier.
En cas d’union consanguine, l’étude des SNP de l’enfant permet d’identifier les régions de son génome pour lesquelles il est homozygote (mêmes SNP hérités de sa mère et de son père). On parle de perte d’hétérozygotie (LOH loss of heterozygosity). Cela permet de mettre en évidence les zones potentiellement à risque de maladies autosomiques récessives. Il s’agit d’une approche spécifique, utilisée lorsque l’on suspecte ce type de transmission.
Études d’association pangénomique (GWAS)
À l’inverse des maladies monogéniques rares, se trouvent les maladies multifactorielles, fréquentes, pour lesquelles il existe un certain degré de prédisposition génétique. Il peut s’agir du diabète, de l’hypertension, de l’infarctus du myocarde…
L’hypothèse ici, est que certains allèles ou certaines combinaisons d’allèles ne sont pas pathogènes en soi, mais prédisposent, favorisent, ou facilitent l’apparition de la maladie. Leur mise en évidence chez un individu ne veut absolument pas dire que la maladie apparaitra (contrairement aux maladies monogéniques bien souvent), mais que l’individu à un risque plus important de présenter ce type de pathologie, par ailleurs fréquente, et qu’il faut d’autant plus la surveiller, la dépister, et la prévenir.
Des études d’association pangénomique (GWAS, genome-wide association studies) sont utilisées pour mettre en évidence ces facteurs favorisants. Il s’agit d’étudier la fréquence de milliers de SNP dans de grandes séries de patients porteurs d’une même pathologie fréquente, en comparaison avec une cohorte de patients sains comparables en tous points par ailleurs (âge, ethnie, mode de vie, sexe…). Le résultat permet en général de mettre en évidence l’association un peu plus fréquente d’un SNP commun avec une maladie commune (effet favorisant) ou l’inverse (effet protecteur). Toutefois ces études ne permettent pas de dire si c’est la mutation du SNP lui-même qui favorise ou protège de la maladie, ou s’il s’agit d’une mutation voisine génétiquement liée au SNP.
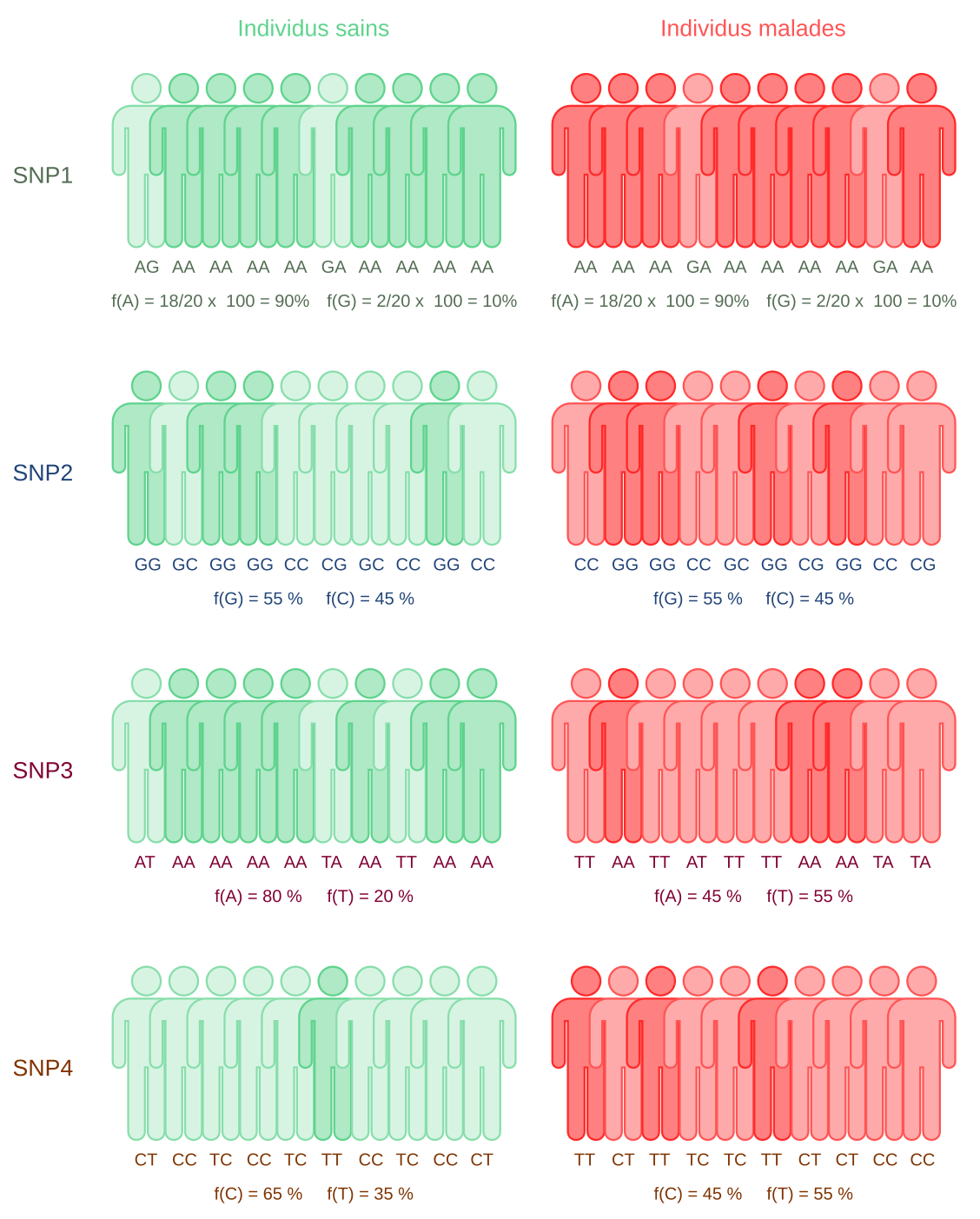
Les études d’association pangénomique consistent à séquencer l’intégralité du génome de plusieurs individus, sains ou malades. Ici, pour simplifier, seuls dix individus de chaque type sont représentés. On compare ensuite les millions de SNP de ces individus. Dans l’exemple présenté ici, seuls quatre SNP sont représentés. Le SNP1 et le SNP2 sont aussi fréquents dans la population saine que dans la population malade. Cela signifie que le gène responsable de la maladie n’est pas situé à proximité de ces SNP. Ils ne présentent donc pas d’intérêt clinique. Pour ces deux SNP, les individus portant au moins un exemplaire de l’allèle le moins fréquent sont représentés de couleur claire, tandis que les homozygotes pour l’allèle le plus fréquent sont en couleur foncée.
Pour le SNP3, la fréquence de l’allèle T est plus importante chez les individus malades que chez les sains. Le gène à l’origine de la maladie est donc proche du SNP3. Les individus portant au moins un exemplaire de l’allèle T sont représentés de couleur claire, les autres de couleur foncée.
Enfin, l’allèle C du SNP 4 est plus présent chez les individus sains que chez les malades. Cela peut signifier que cette version du SNP est liée à un allèle d’un gène qui a un effet protecteur vis-à-vis de la maladie. Les individus portant au moins un exemplaire de l’allèle C sont représentés de couleur claire, les autres de couleur foncée.
Pour le SNP3, on remarque que la plupart des individus malades possèdent l’allèle T (individus en rouge clair) mais pas tous (individus en rouge foncé). De même, pour le SNP4, l’un des individus sains ne possède pas l’allèle C (individu en vert foncé). Cela signifie que la maladie étudiée ici n’est pas monogénique à pénétrance complète. Il s’agit au contraire d’une maladie multifactorielle, avec au moins un gène, à pénétrance incomplète, ayant un effet délétère, et un gène ayant un effet protecteur.
Notons pour finir que, dans les exemples présentés ici, les pourcentages des deux allèles sont soient exactement identiques, soient très différents chez les individus sains et malades. En pratique, les résultats obtenus ne sont pas forcément si extrêmes. Il est donc nécessaire d’employer des tests statistiques pour déterminer la probabilité que les résultats obtenus soient dus au hasard ou non.
Conclusion
Les maladies génétiques monogéniques sont, pour la plupart, aujourd’hui mieux comprises grâce à l’identification des gènes responsables à la fin du XXe siècle. Majoritairement rares, l’identification de la cause de ces pathologies à une époque où la biologie moléculaire n’avait pas la puissance actuelle, a vu l’émergence d’élégantes techniques d’analyses par cartographie génétique sur analyse de cohortes (études de liaison).
À l’heure actuelle, si la tâche semble plus ardue malgré des moyens d’analyses puissants avec des maladies toujours plus rares et des patients uniques, se pose avant tout la question des limites que nous souhaitons franchir dans l’étude génétique de nos populations.
Au-delà de la cartographie des maladies, nous sommes en train de cartographier des facteurs négatifs, comme positifs, mais également ethniques. Les outils d’aujourd’hui permettent le séquençage complet du génome humain en quelques jours. Certaines sociétés privées proposent déjà aux particuliers de séquencer leur ADN et de leur donner ainsi des informations sur leurs ancêtres mais aussi sur leur santé. Est-il souhaitable que des personnes découvrent, sans accompagnement médical, qu’ils sont porteurs de tel ou tel variant génétique impliqué dans une maladie ? Dans la mesure où nous sommes tous porteurs de mutations potentiellement délétères sera-t-il opportun d’avoir recours, en routine, au séquençage du génome des patients ? Faudra-t-il leur communiquer les résultats, notamment ceux en lien avec d’autres pathologies que celle pour laquelle ils sont venus consulter ? Dans un autre registre, faut-il craindre qu’une dérive sélective prénatale se mette en place ? En miroir de ce qui se fait en occident pour la recherche des facteurs d’autisme et de déficience intellectuelle, certaines équipes chinoises ont déjà cartographié les facteurs favorisant un QI élevé.