La maladie de Huntington est une maladie neurologique héréditaire dont les manifestations ont lieu à l’âge adulte. Il n’existe pas de traitement permettant d’en ralentir la progression ou de la guérir. Cette pathologie est due à une mutation dominante dans le gène codant la protéine huntingtine. Cette mutation induit à la fois le gain de nouvelles fonctions toxiques et l’altération des fonctions normales de cette protéine. Une meilleure connaissance de sa fonction et de ses altérations chez les patients et dans des modèles animaux ont permis de mieux comprendre la pathophysiologie de la maladie de Huntington.
C’est un jeune médecin du nom de Georges Huntington qui caractérisa pour la première fois les symptômes de la maladie en 1872. Il souligna la nature héréditaire de la maladie, sa transmission selon un mode dominant et sa manifestation à l’âge adulte. Le travail de Georges Huntington a permis de réhabiliter de nombreux malades pris pour des possédés et persécutés pour sorcellerie ; il a été le point de départ de nombreuses recherches.
La prévalence de la maladie de Huntington est de 2 à 7 pour 100 000 en Europe occidentale [1]. La répartition de cette maladie n’est pas homogène géographiquement. Si certains pays sont très peu touchés par la maladie de Huntington, d’autres régions le sont beaucoup plus. C’est le cas de la région du lac de Maracaibo au Venezuela où la prévalence est de 700 malades pour 100 000 personnes. En France, la maladie touche 6 000 personnes et 12 000 individus sont porteurs de la mutation sans présenter de symptômes (présymptomatiques).
La maladie se déclare généralement à l’âge adulte, en moyenne entre 35 et 45 ans, avec une espérance de vie qui varie entre 15 à 20 ans en moyenne après l’apparition des premiers symptômes. Il existe cependant de rares formes pédiatriques et juvéniles caractérisées par l’apparition des symptômes avant 21 ans.
Description clinique et neuropathologique
La maladie de Huntington est une maladie progressive, caractérisée par une association de trois types de symptômes : moteurs, cognitifs et psychiatriques. Le développement de la maladie est défini par une phase cliniquement silencieuse ou présymptomatique, suivie d’une phase symptomatique pendant laquelle l’état du malade se dégrade progressivement. Dans les premiers stades de la maladie, les patients sont atteints d’anxiété, d’irritabilité ainsi que de dépression. Ils présentent aussi des troubles de la coordination, des mouvements involontaires et des difficultés à planifier. La chorée, signe moteur caractéristique de la maladie de Huntington, devient ensuite plus marquée. La chorée consiste en des mouvements involontaires, touchant aussi bien la racine des membres que leur extrémité, le cou et la face. Les activités volontaires sont de plus en plus difficiles, les troubles de l’articulation de la parole et les sensations de gêne au moment de l’alimentation s’aggravent. Ces symptômes s’accompagnent de changement de la personnalité, avec des comportements agressifs et une désinhibition sociale. Dans les derniers stades de la maladie, les troubles du comportement s’atténuent et les troubles moteurs deviennent sévères. Les malades présentent une perte de poids, des troubles du sommeil et sont généralement totalement dépendants. Ce tableau clinique est très général, il existe une variabilité dans l’apparition et la manifestation des symptômes selon les individus atteints [1].
La lésion caractéristique de la maladie de Huntington est une dégénérescence de certains neurones de deux régions du cerveau, le cortex et le striatum (Figures 1 et 2). La maladie de Huntington conduit à une atrophie sévère du cerveau, celui-ci pouvant perdre jusqu’à 30 % de sa masse [2,3].
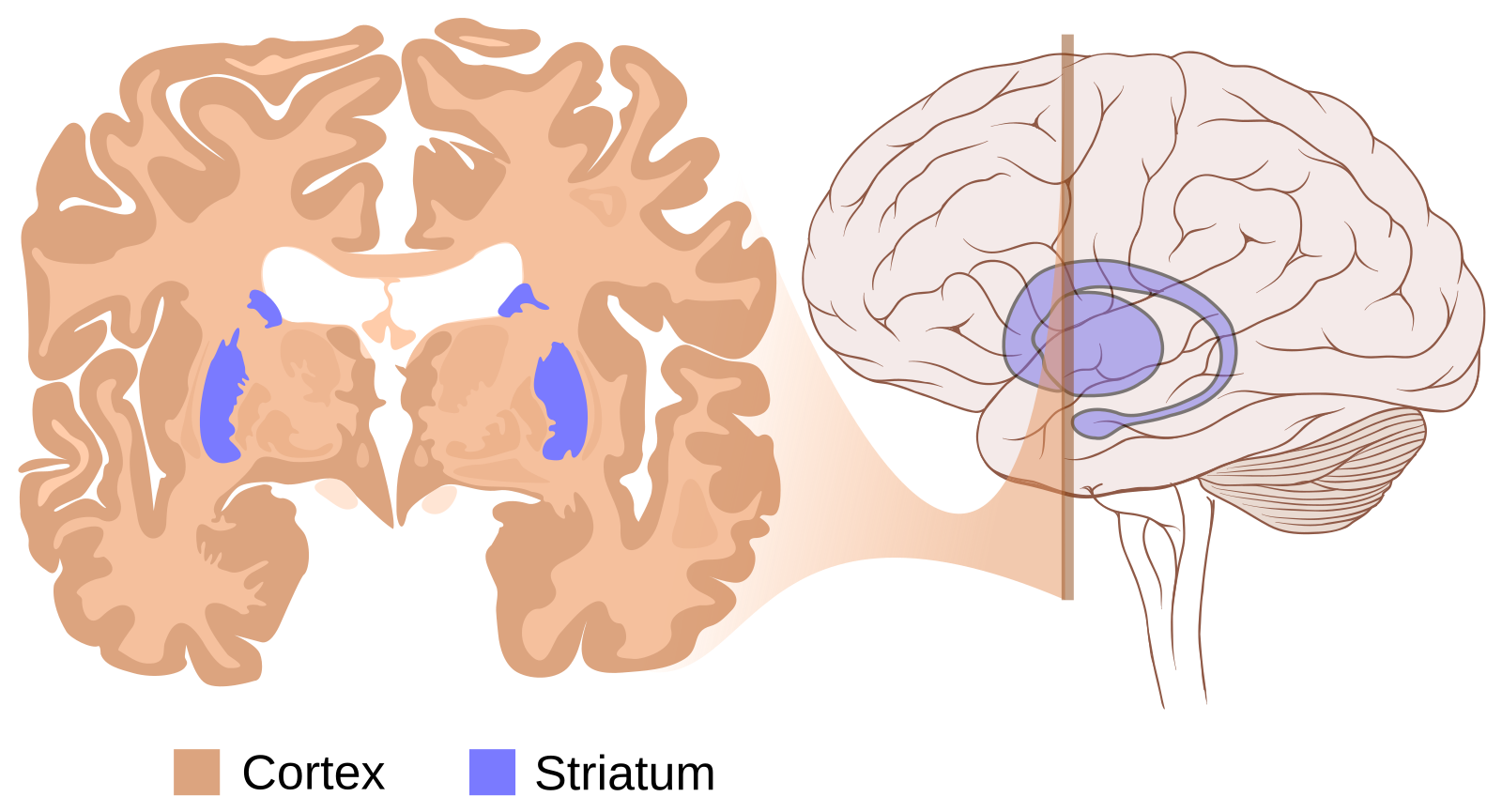
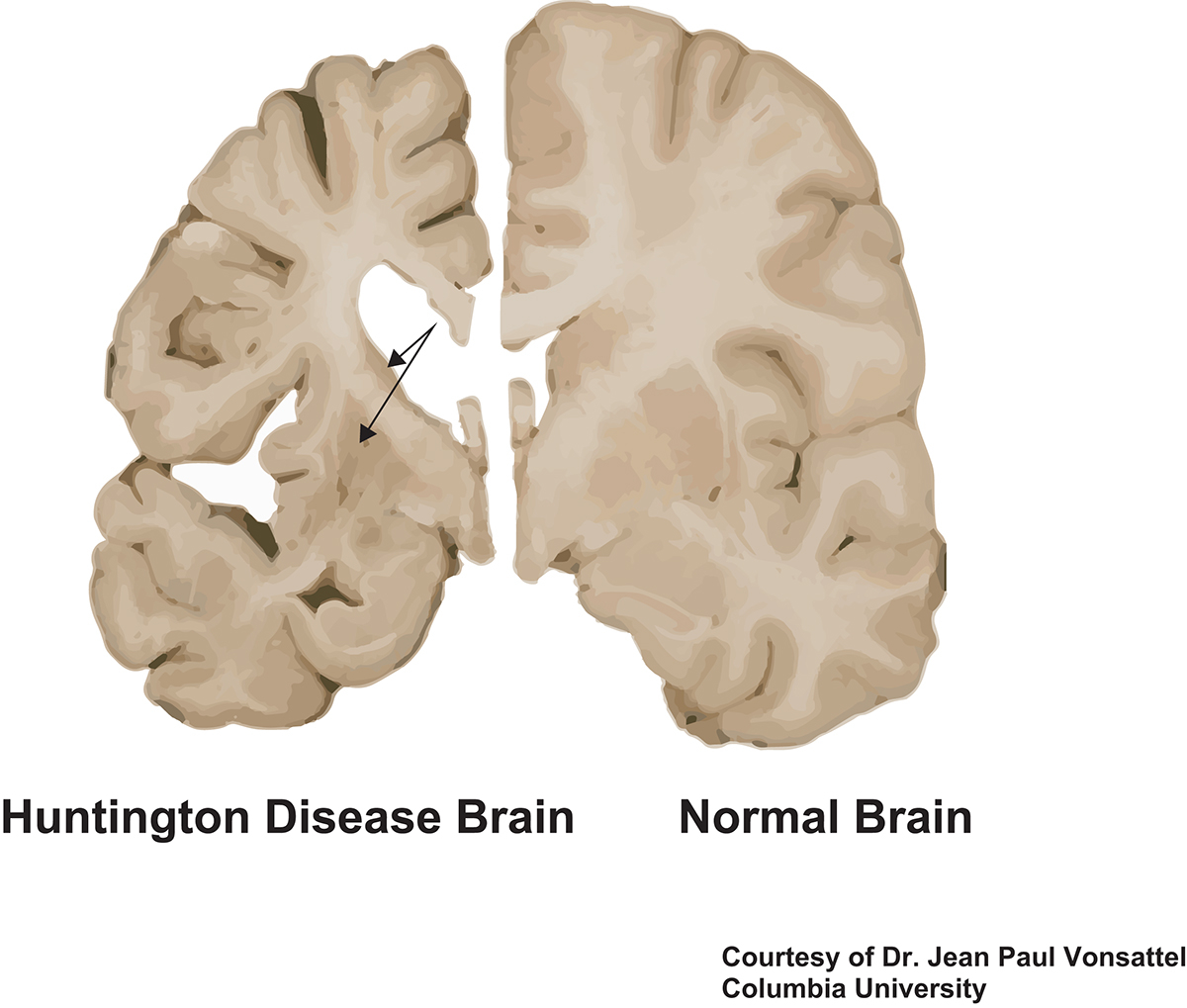
Le cerveau d’un patient atteint de la maladie de Huntington montre des signes d’atrophie, en particulier au niveau du striatum (flèches) et du cortex.
Une maladie génétique
La maladie de Huntington est une maladie génétique autosomale dominante. Le gène huntingtine, ainsi que le type de mutation responsable de la maladie, ont été identifiés en 1993. Ce gène est particulièrement long, avec 67 exons répartis sur 200 kb et code la protéine huntingtine. La mutation localisée dans le premier exon du gène huntingtine correspond à une expansion anormale du triplet de nucléotides CAG dans la région codante. Le triplet CAG code l’acide aminé glutamine (code à une lettre, Q ; code à trois lettres, Gln) à l’extrémité N-terminale de la protéine huntingtine. Les allèles « normaux » du gène huntingtine contiennent de 9 à 35 triplets CAG (codant pour 9 à 35 Q dans la protéine huntingtine) tandis que les allèles à l’origine de la maladie de Huntington présentent un nombre de répétitions supérieur à 36 CAG (>36 Q dans la protéine huntingtine). Les formes adultes de la maladie de Huntington se caractérisent généralement par une expansion comprise entre 40 et 55 CAG, tandis que des répétitions plus longues sont souvent associées à des formes juvéniles [4]. L’instabilité des CAG est liée à des défauts de réplication de l’ADN ainsi qu’à des défauts de réponse aux dommages de l’ADN.
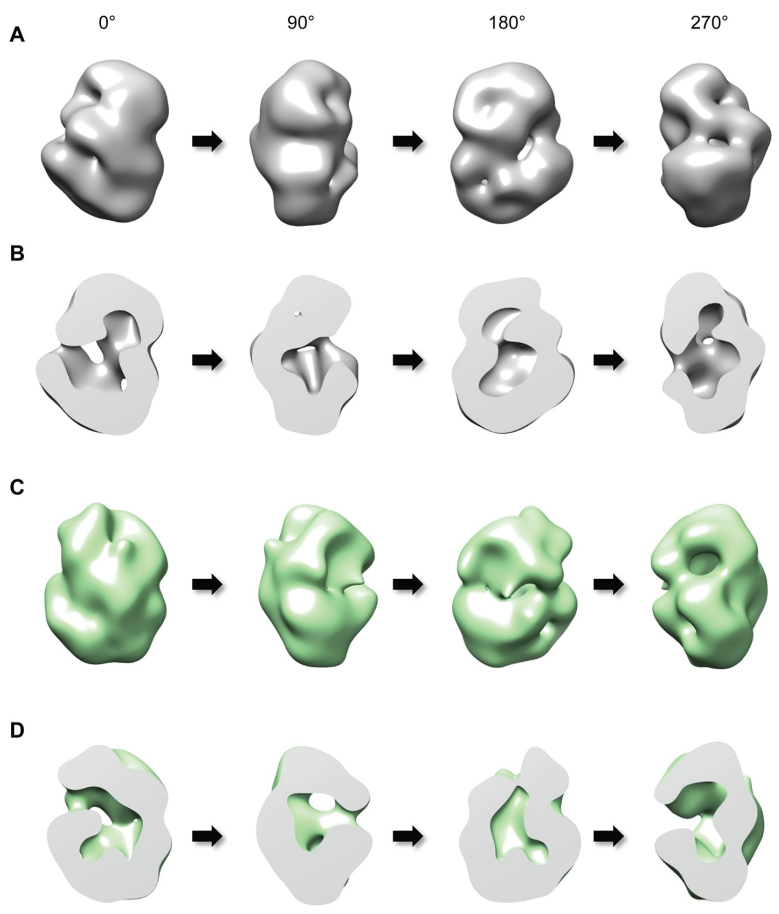
Vues externes (A) et en coupe (B) de la huntingtine Q23 (avec 23 glutamines au niveau de l’extrémité N-terminale, situation physiologique) selon différents angles autour de l’axe vertical.
Les lignes C et D montrent les résultats obtenus pour la huntingtine Q78 (avec une expansion anormale de 78 glutamines au niveau de l’extrémité N-terminale, situation pathologique). On remarque que la forme de la protéine est altérée par rapport à la huntingtine Q23.
Ces résultats ont été obtenus par cryo-microscopie électronique.
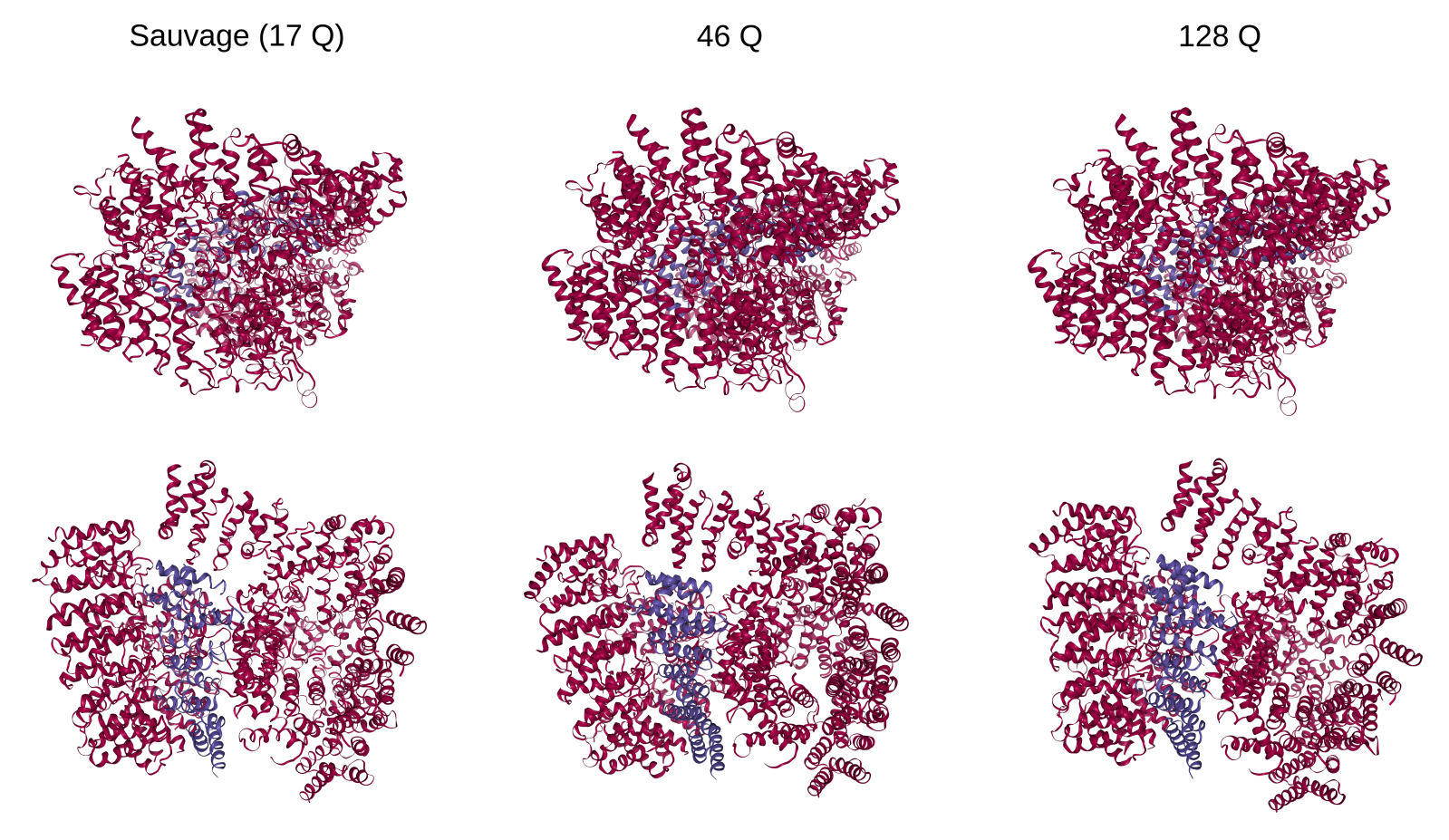
La huntingtine peut se lier à la protéine HAP40 (HTT-associated protein 40). À l’inverse de ce que l’on observe pour la huntingtine seule (voir figure 3), la structure du complexe huntingtine-HAP40 semble peu affectée par le nombre de glutamines en position N-terminale (17 : situation physiologique ; 46 et 128 : situations pathologiques). La structure de ces complexes a été déterminée par cryo-microscopie électronique.
Les fichiers de molécules sont disponibles sur le site RSCB PDB : huntingtine 17Q (6EZ8), 46Q (7DXJ) et 128Q (7DXK).
Publications de référence : huntingtine 17Q : Guo et coll., 2018, Nature ; huntingtines 46Q et 128Q : Huang et coll., 2021, Structure.
La huntingtine est une protéine de 350 kilodaltons (kDa) ne possédant pas d’homologie particulière avec d’autres protéines connues [5]. Elle a une expression ubiquitaire, avec des niveaux plus élevés dans le cerveau et les testicules. Dans le cerveau, elle est présente à des niveaux équivalents dans de nombreuses structures, ce qui ne permet pas d’expliquer pourquoi le cortex et le striatum sont particulièrement touchés lors de la progression pathologique. À l’intérieur des cellules, la huntingtine est trouvée dans le cytoplasme, dans les neurites (axones et dendrites) et, en particulier, au niveau des synapses. Elle s’associe avec différents organites et structures, comme le compartiment endoplasmique, les mitochondries, des vésicules, les microtubules et la membrane plasmique. Bien que majoritairement cytoplasmique, la huntingtine est aussi trouvée dans le noyau. Étant donné sa localisation subcellulaire, elle apparaît impliquée dans de nombreuses fonctions cytoplasmiques et nucléaires. Ceci est confirmé par le fait que la huntingtine interagit avec des protéines variées impliquées dans l’expression des gènes, le transport intracellulaire, la signalisation et le métabolisme [5].
Pendant longtemps, le gain de nouvelles fonctions toxiques par la huntingtine mutante a été la voie étudiée de façon préférentielle pour comprendre les mécanismes mis en jeu dans la maladie de Huntington. Ceci était lié au fait que cette maladie possède un mode de transmission dominant, les individus hétérozygotes et homozygotes pour la mutation ayant des pathologies similaires. Les gains de fonctions toxiques correspondent notamment à des repliements anormaux de la huntingtine mutante (Figure 3) et à ses interactions anormales avec d’autres protéines. À l’heure actuelle, de plus en plus d’études soulignent qu’en parallèle à ce gain de nouvelles fonctions toxiques, les modifications des fonctions normales de la huntingtine jouent également un rôle central dans la maladie. Perte et gain de fonctions peuvent affecter les mêmes mécanismes et sont ainsi à prendre en compte pour comprendre comment la huntingtine mutée conduit à la mort neuronale [5].
Ainsi, pour étudier les gains de fonctions de toxiques, de nombreux travaux se sont intéressés aux mécanismes conduisant à la dysfonction et à la mort neuronale lorsque la huntingtine mutante est exprimée dans des modèles cellulaires et animaux (voir paragraphe suivant). Un autre axe de recherche a été de caractériser les fonctions la protéine normale, puis d’analyser ces fonctions lorsque la huntingtine est mutée comme dans la maladie de Huntington. Ces travaux ont montré que la huntingtine occupe des fonctions cellulaires centrales qui ont pour conséquences physiologiques de permettre le développement de l’organisme en général (et plus particulièrement du cerveau) et de maintenir le fonctionnement et la survie neuronale dans le cerveau adulte. La huntingtine est importante pour le transport intracellulaire de vésicules le long des microtubules ainsi que pour d’autres dynamiques intracellulaires telles que l’endocytose et le recyclage de vésicules. L’expression de la huntingtine n’est pas restreinte aux neurones post-mitotiques et elle régule la division cellulaire. Elle est également importante pour l’assemblage et le fonctionnement des cils, notamment le cil primaire des neurones, qui est un organite impliqué dans la réception de signaux extracellulaire et leur transduction. Lorsque la huntingtine est mutée, toutes les fonctions caractérisées à ce jour sont altérées [5].
Afin d’illustrer cette notion de fonction/dysfonction, nous citerons l’exemple du transport du facteur trophique BDNF, brain-derived neurotrophic factor. Ce facteur est absolument requis par le striatum pour son développement et sa survie ; or il est peu produit par les neurones du striatum. La majorité du BDNF qui arrive au striatum provient des neurones du cortex : il y est produit et transporté par un mécanisme qui implique la huntingtine (Figure 5). Lorsque celle-ci est mutée, ce transport se fait moins bien, ce qui réduit l’apport en BDNF vers le striatum conduisant à une sensibilité accrue de ces neurones à la mort [6]. Ainsi, une meilleure compréhension des fonctions normales de la huntingtine peut permettre de mieux comprendre les mécanismes pathologiques mis en jeu dans la maladie de Huntington. On peut également – comme nous le verrons dans la dernière partie – imaginer des pistes thérapeutiques pour la maladie de Huntington, qui consisteraient à rétablir les dysfonctions de la huntingtine.
En contexte physiologie, la huntingtine sauvage s’assemble avec d’autres protéines (dynéine, dynactine, kinésine et HAP1) qui permettent le transport de vésicules contenant du BDNF le long des microtubules. Le BDNF est ainsi transporté des neurones du cortex aux neurones du striatum et permet la survie de ces derniers.
Dans le cas de la maladie de Huntington, le transport de vésicules de BDNF est altéré, ce qui compromet la survie des neurones du striatum.
Une cascade d’évènements pathologiques
Historiquement, la plupart des études se sont concentrées sur l’identification des conséquences cellulaires de l’expression de la huntingtine mutante dans des lignées cellulaires, des cultures primaires de neurones et des modèles animaux. Cela a permis la description d’une cascade d’événements cellulaires qui conduisent finalement à la dysfonction et la perte neuronale (Figure 6). Le clivage protéolytique de la huntingtine mutante en fragments et la translocation de ces fragments vers le noyau est une étape clé dans la maladie [7]. La partie N-terminale des fragments générés contient la mutation ; ces fragments sont d’autant plus toxiques qu’ils sont courts. Le clivage et la translocation nucléaire sont tous deux absolument nécessaires pour induire la mort neuronale [7,8]. Plusieurs protéases peuvent cliver la huntingtine et différents produits de clivage ont été trouvés dans le cerveau des patients et dans des modèles murins. L’activation des protéases pourrait résulter de diverses stress subis par les neurones tels que des niveaux excessifs de calcium cytosolique (excitotoxicité) et l’activation de la machinerie apoptotique [9]. Une fois dans le noyau, la huntingtine mutante induit la mort des neurones par la dérégulation de la machinerie transcriptionnelle, en particulier en interférant avec les facteurs de transcription [8,10]. En bon accord, l’analyse du profil global d’expression des gènes révèle que plusieurs catégories fonctionnelles de gènes ont leurs niveaux modifiés par la huntingtine mutante [10,11].
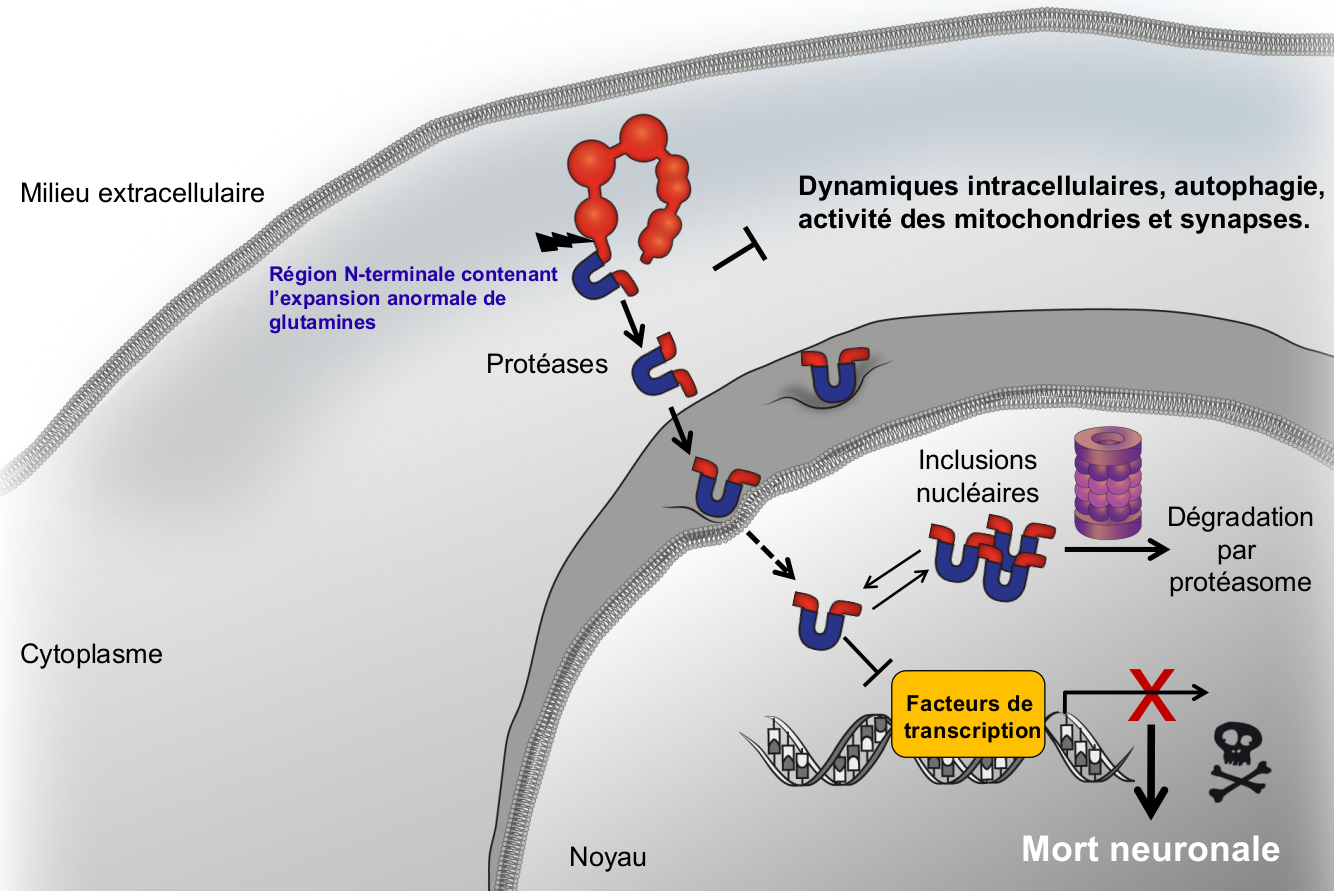
La huntingtine mutante est clivée en fragments qui transloquent vers le noyau, où ils induisent la mort neuronale par la dérégulation de la machinerie de transcription. Les fragments peuvent former des agrégats (inclusions nucléaires) qui seront dégradés par la voie du protéasome ou qui s’accumuleront, une des caractéristiques de la maladie de Huntington. En parallèle, la huntingtine mutante et ses fragments conduisent à de multiples déficiences par exemple dans la régulation des dynamiques intracellulaires, de l’autophagie, de l’activité des mitochondries et des synapses.
En plus de cette voie canonique, plusieurs effets de la huntingtine mutante et de ses fragments ont été décrits, notamment : l’inhibition du protéasome (impliqué dans la dégradation des protéines) et de l’autophagie (mécanisme d’autodigestion d’une partie du cytosol ou des organites) [12,13] ; des anomalies mitochondriales et des déficiences métaboliques associées à ces anomalies ; l’altération de différentes dynamiques intracellulaires dont l’endocytose et le transport le long des microtubules ; des déficiences de l’activité des synapses ; l’altération de la signalisation calcique et l'excitotoxicité causée par une libération accrue de glutamate et le dysfonctionnement de plusieurs neurotransmetteurs [5].
Enfin, comme pour plusieurs autres maladies neurodégénératives telles que les maladies d’Alzheimer et de Parkinson, une des caractéristiques de la maladie de Huntington est l’agrégation de la huntingtine mutante au niveau nucléaire et cytoplasmique [14]. Le rôle exact de ces agrégats reste sujet à controverse. Si les agrégats ont été initialement décrits comme étant les espèces toxiques dans la maladie de Huntington, des études plus récentes ont montré qu’il n’y a pas de corrélation directe entre les agrégats et la mort neuronale. Les agrégats pourraient au contraire avoir un effet bénéfique, car ils représenteraient initialement un mécanisme de protection de la cellule pour stocker temporairement les formes solubles de la huntingtine mutantes qui elles sont très toxiques [8,15]. Ainsi, l’effet des agrégats pourrait dépendre du stade de la maladie, de leur nature et de leur localisation subcellulaire.
Quelles pistes thérapeutiques ?
Il n’existe pour l’instant pas de solution thérapeutique permettant de retarder le développement de la maladie de Huntington ou d’en guérir. Les patients sont pris en charge par plusieurs spécialistes de la santé : médecins et professionnels paramédicaux. Seuls les symptômes sont traités, avec des médicaments qui ne sont donc pas spécifiques de la maladie de Huntington, tels que des neuroleptiques et des antidépresseurs. Au-delà du coût financier de la prise en charge des patients, la maladie de Huntington entraîne des drames personnels vécus par les conjoints et les familles nécessitant une prise en charge psychologique, psychiatrique et sociale.
Ainsi, une solution thérapeutique est absolument nécessaire pour cette pathologie et fait l’objet d’intenses recherches par de nombreuses équipes dans le monde depuis la découverte du gène en 1993. Au cours des dernières années, de nombreuses pistes thérapeutiques ont été suivies mais sans donner de résultats probants. Ces pistes incluent la recherche d’inhibiteurs de l’agrégation de la huntingtine mutante ou de molécules réduisant le stress oxydatif. La maladie de Huntington étant caractérisée par des déficits énergétiques et des défauts au niveau des mitochondries, des molécules stimulant la production d’ATP et l’activité mitochondriale, comme la créatine ou la coenzyme Q10, ont été testées chez les patients, mais à nouveau sans effets bénéfiques.
Comme nous l’avons mentionné, une des caractéristiques de la maladie est la réduction de l’apport du facteur neurotrophique BDNF jusqu’au striatum. Cependant, les stratégies visant à rétablir les niveaux cérébraux de la protéine BDNF via des greffes de cellules productrices ou par transduction virale du gène BDNF dans les neurones n’ont pas montré d’efficacité. Ces résultats négatifs pourraient être dus à la nécessité que le BDNF soit exprimé dans les bons circuits neuronaux pour être actif et non pas produit de manière non sélective dans l’ensemble du cerveau. Un certain nombre de molécules ayant potentiellement des effets neuroprotecteurs en promouvant la production de BDNF sont actuellement en développement.
Au-delà de ces solutions de pharmacologie classique, le développement d’oligonucléotides antisens est en plein essor. En ciblant directement les ARN messagers codant la huntingtine, ceux-ci conduisent à l’inhibition de la production de la huntingtine mutante. En particulier, la stratégie développée par IONIS/Roche permet de diminuer efficacement l’expression de la huntingtine mutante [16]. Cependant, l’essai de phase 3 sur plus de 800 patients a récemment été stoppé suite à une aggravation de l’état des patients et à un rapport risque/bénéfice en défaveur des patients [17]. Cet échec thérapeutique est probablement lié au fait que cette approche n’est pas spécifique de l’allèle muté mais diminue également, chez les patients hétérozygotes, l’expression de la huntingtine sauvage fonctionnelle. En effet, comme expliqué plus haut, la huntingtine sauvage possède des propriétés neuroprotectrices bénéfiques pour le cerveau, que ce soit au cours du développement ou chez l’adulte [5]. Il faut également noter que ces oligonucléotides (RG6042 ou Tominersen) nécessitent d’être administrés à plusieurs reprises au patient par la voie intrathécale, c’est-à-dire dans l’espace sous-arachnoïdien afin d’atteindre le liquide céphalo-rachidien, ce qui est très contraignant. Une nouvelle stratégie développée par la société Wave Life Sciences vise à développer des molécules antisens sélectives de l’allèle muté. Cette stratégie semble plus prometteuse que celle développée par Roche mais reste coûteuse et nécessitera des injections intrathécales répétées. De plus, même si les molécules antisens sont spécifiques de l’allèle muté, les patients hétérozygotes ne produiront plus qu’une demi-dose de la huntingtine ce qui potentiellement présente un risque. En effet, comme nous l’avons vu plus haut, la huntingtine joue un rôle central dans des fonctions cellulaires importantes et, dans des modèles animaux, l’expression d’une demi-dose de huntingtine suffit à altérer ces fonctions. Enfin, il faut noter que tous les patients ne pourront prétendre au traitement car cette molécule antisens cible un polymorphisme nucléotidique tous ne présentent pas.
En conclusion, la maladie de Huntington est une maladie neurologique héréditaire grave pour laquelle il n’existe pas de solution thérapeutique à l’heure actuelle. L’identification et le développement de nouvelles approches thérapeutiques ne se fera pas sans une meilleure connaissance fondamentale des mécanismes physiopathologiques à l’origine de cette maladie.
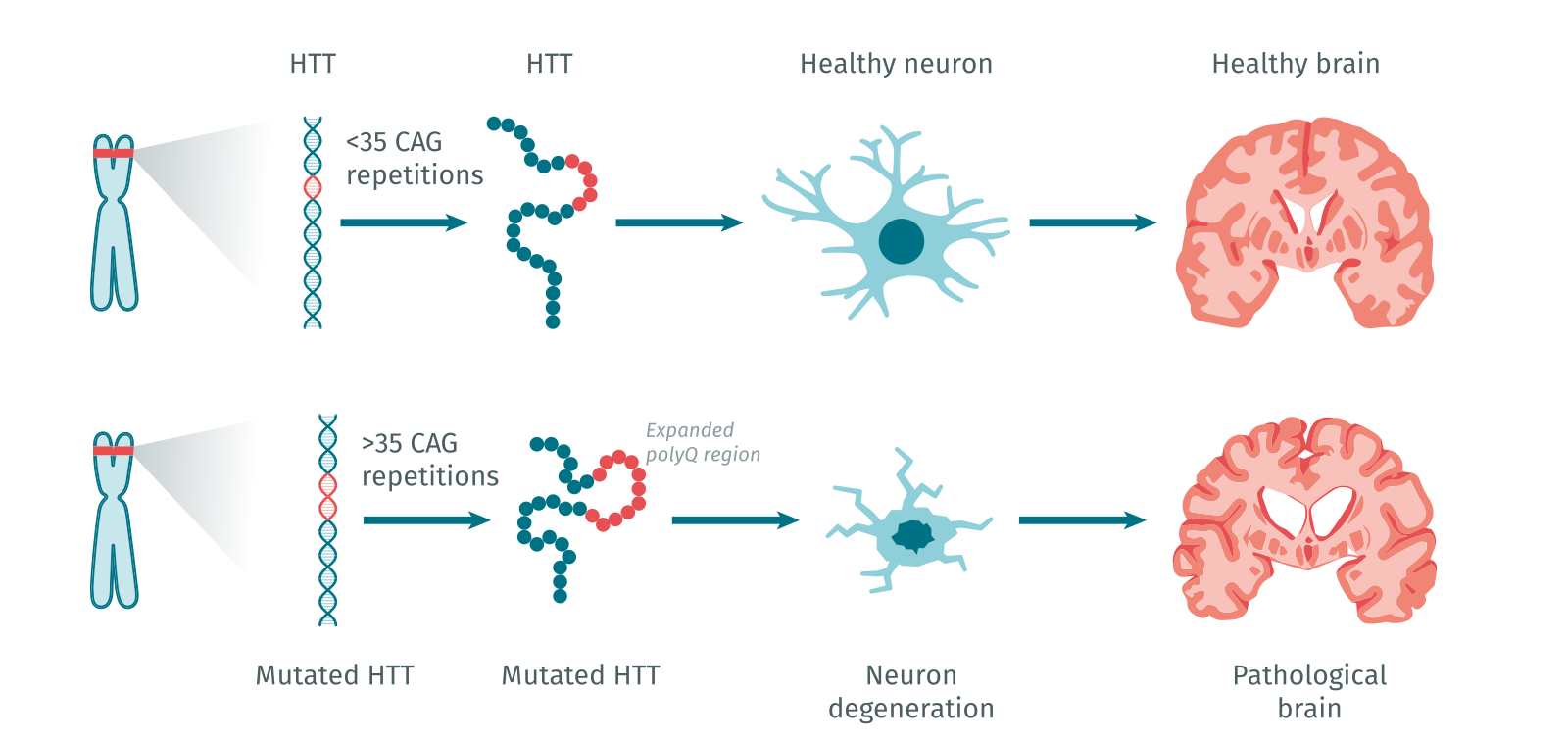
La maladie de Huntington est une maladie génétique dominante due à la mutation du gène huntingtine. Ce gène contient une expansion du triplet de nucléotides CAG dans sa région codante, conduisant à une expansion de l’acide aminé glutamine dans la protéine huntingtine. Cette expansion est anormalement étendue dans la maladie. Les allèles « normaux » du gène huntingtine contiennent de <35 triplets CAG (<35Q), les allèles à l’origine de la maladie de Huntington sont >36 CAG (>36Q).
Références
- C. A. Ross, S. J. Tabrizi, Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol10, 83-98 (2011).
- S. J. Tabrizi, R. I. Scahill, A. Durr, R. A. Roos, B. R. Leavitt, R. Jones, G. B. Landwehrmeyer, N. C. Fox, H. Johnson, S. L. Hicks, C. Kennard, D. Craufurd, C. Frost, D. R. Langbehn, R. Reilmann, J. C. Stout, T.-H. Investigators, Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol10, 31-42 (2011).
- H. D. Rosas, S. Y. Lee, A. C. Bender, A. K. Zaleta, M. Vangel, P. Yu, B. Fischl, V. Pappu, C. Onorato, J. H. Cha, D. H. Salat, S. M. Hersch, Altered white matter microstructure in the corpus callosum in Huntington's disease: implications for cortical "disconnection". Neuroimage49, 2995-3004 (2010).
- HDCRG, A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell72, 971-983 (1993).
- F. Saudou, S. Humbert, The Biology of Huntingtin. Neuron89, 910-926 (2016).
- H. Vitet, V. Brandt, F. Saudou, Traffic signaling: new functions of huntingtin and axonal transport in neurological disease. Curr Opin Neurobiol63, 122-130 (2020).
- R. K. Graham, Y. Deng, E. J. Slow, B. Haigh, N. Bissada, G. Lu, J. Pearson, J. Shehadeh, L. Bertram, Z. Murphy, S. C. Warby, C. N. Doty, S. Roy, C. L. Wellington, B. R. Leavitt, L. A. Raymond, D. W. Nicholson, M. R. Hayden, Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell125, 1179-1191 (2006).
- F. Saudou, S. Finkbeiner, D. Devys, M. E. Greenberg, Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell95, 55-66 (1998).
- M. P. Parsons, L. A. Raymond, Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron82, 279-293 (2014).
- L. Cui, H. Jeong, F. Borovecki, C. N. Parkhurst, N. Tanese, D. Krainc, Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell127, 59-69 (2006).
- P. Langfelder, J. P. Cantle, D. Chatzopoulou, N. Wang, F. Gao, I. Al-Ramahi, X. H. Lu, E. M. Ramos, K. El-Zein, Y. Zhao, S. Deverasetty, A. Tebbe, C. Schaab, D. J. Lavery, D. Howland, S. Kwak, J. Botas, J. S. Aaronson, J. Rosinski, G. Coppola, S. Horvath, X. W. Yang, Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci19, 623-633 (2016).
- A. L. Goldberg, Protein degradation and protection against misfolded or damaged proteins. Nature426, 895-899 (2003).
- Y. N. Rui, Z. Xu, B. Patel, Z. Chen, D. Chen, A. Tito, G. David, Y. Sun, E. F. Stimming, H. J. Bellen, A. M. Cuervo, S. Zhang, Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol17, 262-275 (2015).
- M. DiFiglia, E. Sapp, K. O. Chase, S. W. Davies, G. P. Bates, J. P. Vonsattel, N. Aronin, Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science277, 1990-1993 (1997).
- M. Arrasate, S. Mitra, E. S. Schweitzer, M. R. Segal, S. Finkbeiner, Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature431, 805-810 (2004).
- S. J. Tabrizi, B. R. Leavitt, G. B. Landwehrmeyer, E. J. Wild, C. Saft, R. A. Barker, N. F. Blair, D. Craufurd, J. Priller, H. Rickards, A. Rosser, H. B. Kordasiewicz, C. Czech, E. E. Swayze, D. A. Norris, T. Baumann, I. Gerlach, S. A. Schobel, E. Paz, A. V. Smith, C. F. Bennett, R. M. Lane, I.-H. S. S. T. Phase 1-2a, Targeting Huntingtin Expression in Patients with Huntington's Disease. N Engl J Med380, 2307-2316 (2019).
- D. Kwon, Failure of genetic therapies for Huntington's devastates community. Nature593, 180 (2021).
- R. Jung, Y. Lee, D. Barker, K. Correia, B. Shin, J. Loupe, R. L. Collins, D. Lucente, J. Ruliera, T. Gillis, J. S. Mysore, L. Rodan, J. Picker, J. M. Lee, D. Howland, R. Lee, S. Kwak, M. E. MacDonald, J. F. Gusella, I. S. Seong, Mutations causing Lopes-Maciel-Rodan Syndrome are huntingtin hypomorphs. Hum Mol Genet 30, 135-148 (2021).