

Des technologies qui permettent de déterminer la succession des nucléotides, c’est-à-dire la séquence d’une molécule d’acide nucléique (ADN ou ARN) existent depuis les années 70. La méthode Sanger, la plus utilisée, a permis le séquençage de divers génomes dont celui de l’être humain, et a ainsi révolutionné la génomique et la biologie de manière générale. Toutefois, le séquençage du génome humain avec la technologie Sanger a été un immense effort qui a pris plus de dix ans et a coûté environ trois milliards de dollars [1]. Il est donc évident que cette méthode n’est pas adaptée au séquençage de grands génomes, et c’est pour cette raison que de nouvelles technologies ont été développées. Dans cet article nous discutons de ces méthodes dites « séquençage à haut débit » ou « de nouvelle génération » et qui ont révolutionné la génomique en permettant de séquencer un génome humain en quelques jours pour moins de 1000 dollars. Nous présenterons également les technologies de troisième génération, encore plus récentes, qui, depuis quelques années, constituent une nouvelle révolution.
La méthode Sanger
La technologie Sanger a été décrite dans les articles Le séquençage d’un ADN et Le séquençage des génomes. Ici nous résumerons de façon succincte le principe de cette méthode. Le séquençage Sanger d’un fragment d’ADN nécessite d’abord de le cloner dans un plasmide, qui est ensuite introduit dans une cellule hôte, en général une bactérie ou une levure (Figure 1). En se multipliant, cette cellule hôte produit un grand nombre de copies de chaque fragment d’ADN d’origine. Après purification, cet ADN peut être séquencé en utilisant une polymérase qui synthétise un brin complémentaire à partir d’un brin matrice du fragment d’ADN d’intérêt ; quand la polymérase incorpore un des quatre « didésoxynucléotides » (ddATP, ddCTP, ddGTP, ou ddTTP présents séparément dans quatre réactions individuelles), la synthèse s’arrête. Cela génère un mélange de molécules qui se terminent à chaque position où se trouve un A, un C, un G, ou un T (selon le type de didésoxynucléotide présent). Les fragments dans ce mélange sont séparés selon leur taille par électrophorèse sur gel. La connaissance du didésoxynucléotide qui a été incorporé dans chaque réaction permet ainsi de déduire la séquence du fragment d’ADN d’intérêt.
La naissance des méthodes de séquençage de nouvelle génération
Après la complétion du projet de séquençage du génome humain (Human Genome Project, HGP), l’ambition était de séquencer un grand nombre de génomes afin d’étudier la variation génétique et de réaliser des études d’association pangénomique (genome-wide association studies – GWAS), dans lesquelles on essaye d’identifier des liens entre des maladies génétiques et des profils génétiques spécifiques. Dans ce but, des méthodes de séquençage de nouvelle génération (next-generation sequencing, NGS) ont été développées. Ces méthodes partagent trois améliorations majeures par rapport au séquençage Sanger :
- Au lieu d’un clonage moléculaire des fragments d’ADN suivi par l’introduction dans des cellules hôtes et l’isolement de chaque clone individuellement, une banque qui contient l’ensemble des fragments est faite directement dans un tube (in vitro). La Figure 1 montre de façon schématique la procédure ; des fragments d’ADN, générés par coupures aléatoires (enzymatiques ou mécaniques) de l’ADN génomique, sont reliés à des petites molécules d’ADN de séquences connues appelés « adaptateurs ». Les coupures aléatoires génèrent des fragments d’une grande diversité de tailles (entre environ 50 nt et 2000 nt). Une sélection de taille est généralement effectuée pour deux raisons : (1) éliminer les fragments plus courts que la longueur de séquençage, et (2) éliminer les fragments trop longs (d’une taille supérieure à environ 1000 nt). Cette dernière étape est importante pour les techniques de NGS qui nécessitent ensuite une amplification par PCR (polymerase chain reaction), qui est moins efficace sur de longs fragments. Notez bien que tandis que dans la Figure 1, une seule molécule d’ADN génomique est montrée, en réalité une banque est faite à partir d’un grand nombre de copies des molécules d’ADN génomique à séquencer1.
- Alors que les machines développées pour le projet de séquençage du génome humain n’étaient capables d’effectuer que quelques centaines de réactions de séquençage Sanger en parallèle, les séquenceurs NGS peuvent faire des millions voire des milliards de réactions de séquençage en parallèle.
- Les technologies NGS ne nécessitent pas de séparation des fragments par électrophorèse ; la détection des nucléotides incorporés par la polymérase est faite directement après chaque cycle d’incorporation.
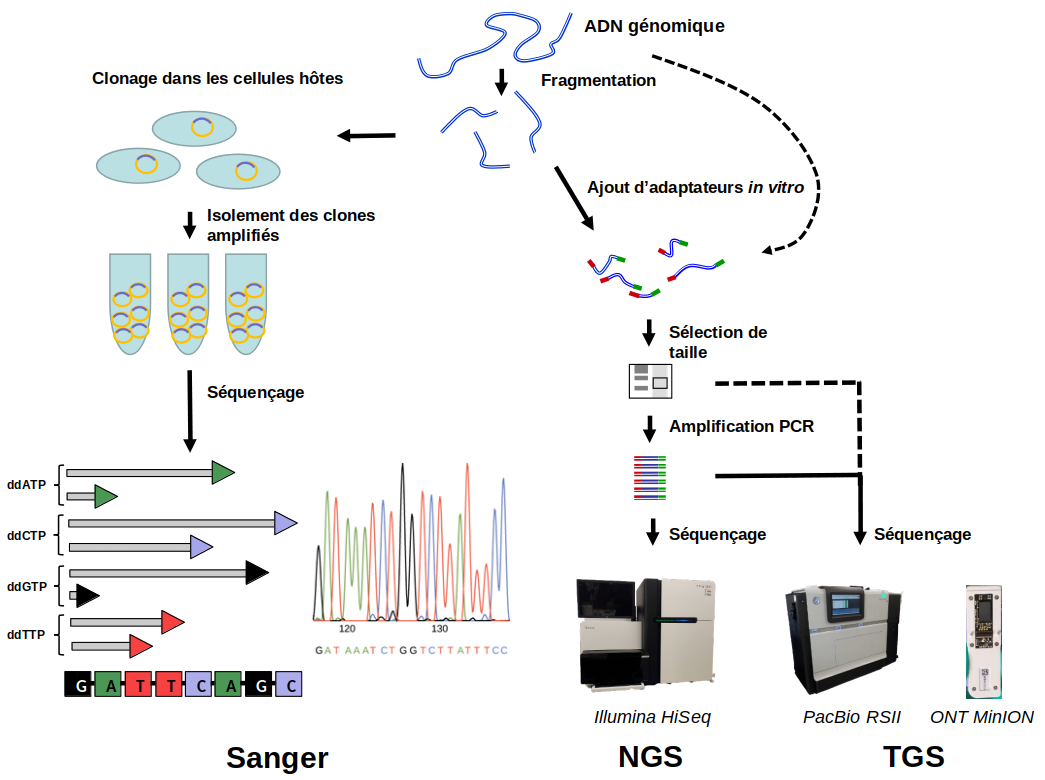
Les préparations des banques NGS et TGS suivent globalement les mêmes procédures, sauf que pour le TGS les étapes de fragmentation de l’ADN génomique et d’amplification par PCR sont optionnelles (indiqué par flèches interrompues). Les adaptateurs sont indiqués en rouge et vert.
Crédits images : séquençage Sanger : OpenStax, CC BY ; Illumina HiSeq 2500 : Konrad Förstner, CC0, Wikimedia ; PacBio RSII : Konrad Förstner, CC BY, Flickr ; ONT MinION : cirosantilli2, CC BY-SA, Wikimedia.
La première technologie NGS était la méthode « 454 », lancée en 2005 par la société 454 Life Sciences. Leur séquenceur 454 Genome Sequencer produisait environ 200 000 lectures d’une longueur de 110 paires de bases par run, c’est-à-dire par cycle de fonctionnement. Un an plus tard, la technologie de Solexa (maintenant Illumina) apparaissait, suivie par SOLiD (Sequencing by Oligo Ligation Detection), commercialisée par Life Technologies en 2007. Les premiers séquenceurs Illumina et SOLiD produisaient plusieurs dizaines de millions de lectures, donc beaucoup plus que la machine 454. En revanche, ces lectures étaient beaucoup plus courtes, environ 35 pb seulement. Une comparaison plus complète de ces trois technologies, qui prend également en compte les temps de runs et leurs coûts a été présenté par Mardis (2008) [2]. Trois ans plus tard, en 2010, Ion Torrent apparaissait, une technologie à base de semi-conducteurs, sans détection optique des nucléotides fluorescents. Cette technologie était moins chère et plus rapide que les autres. La première machine Ion Torrent produisait environ 3 millions de séquences d’une longueur de 100 nucléotides maximum.
Au début, ces différentes technologies étaient en concurrence, mais grâce à une évolution plus rapide de la qualité des données, du débit et de la longueur de lectures que les autres technologies, Illumina a progressivement renforcé sa position sur le marché et a aujourd’hui un quasi-monopole.
Comment fonctionne la technologie Illumina ?
Illumina est aujourd’hui la technologie la plus largement utilisée ; nous discuterons donc seulement de cette technologie, sans rentrer dans les détails des autres méthodes. Pour une description détaillée des diverses technologies, voir [3]. Comme mentionné ci-dessus, les molécules d’une banque NGS contiennent des adaptateurs aux extrémités, qui ont deux fonctions principales : (1) l’accrochage et l’amplification sur une cellule de flux (flow cell), qui est une plaque en verre où les réactions de séquençage et la visualisation des nucléotides incorporés ont lieu, et (2) l’hybridation d’une amorce qui sert de point de départ pour le séquençage. La Figure 2A montre schématiquement l’amplification des molécules de la banque sur une cellule de flux Illumina. Les fragments d’ADN à séquencer sont d’abord dénaturés. Les molécules simple brin obtenues vont alors s’hybrider à différents endroits de la cellule de flux, grâce à la complémentarité de leurs adaptateurs avec les oligonucléotides attachés de façon covalente à la cellule de flux (①). Suivons le devenir de l’une de ces molécules d’ADN. Un brin complémentaire au brin matrice est synthétisé par une polymérase (②). Ensuite, une étape de dénaturation sépare le brin matrice d’origine et le brin complémentaire néosynthétisé. Après une étape de lavage, qui permet d’éliminer le brin matrice d’origine (③), l’extrémité libre du brin complémentaire s’hybride à un oligonucléotide complémentaire fixé à proximité sur la cellule de flux (④). Cela permet une nouvelle synthèse d’un brin complémentaire (et donc identique au brin matrice d’origine, ⑤), suivie par une nouvelle étape de dénaturation afin de séparer les deux brins. On a alors les deux brins côte à côte sur la cellule de flux (⑥). Le même processus se répète un certain nombre de fois (⑦,⑧), ce qui aboutit à la formation d’un « cluster », un groupe d’un millier de molécules identiques au brin matrice d’origine. Ce processus est connu sous le nom d’amplification en pont (bridge amplification) dans la littérature. À d’autres endroits de la cellule de flux, se trouvent d’autres clusters correspondant à l’amplification d’autres fragments de la banque d’ADN à séquencer (Figure 2B). Cette étape d'amplification est nécessaire à l'obtention d'un signal suffisamment important au moment du séquençage. À l'issue de cette étape, des amorces de séquençage s’hybrident à tous les brins de tous les clusters, et servent de point de démarrage du séquençage. Un seul nucléotide marqué est incorporé à chaque cycle, suivi par une étape d’imagerie afin de déterminer quel nucléotide a été incorporé dans chaque cluster (Figure 2C).
Au fil des années, Illumina a agrandi sa gamme de séquenceurs et propose aujourd’hui des machines qui peuvent générer de 4 millions à 20 milliards de séquences par run, avec une longueur maximum de 150 à 300 pb1.
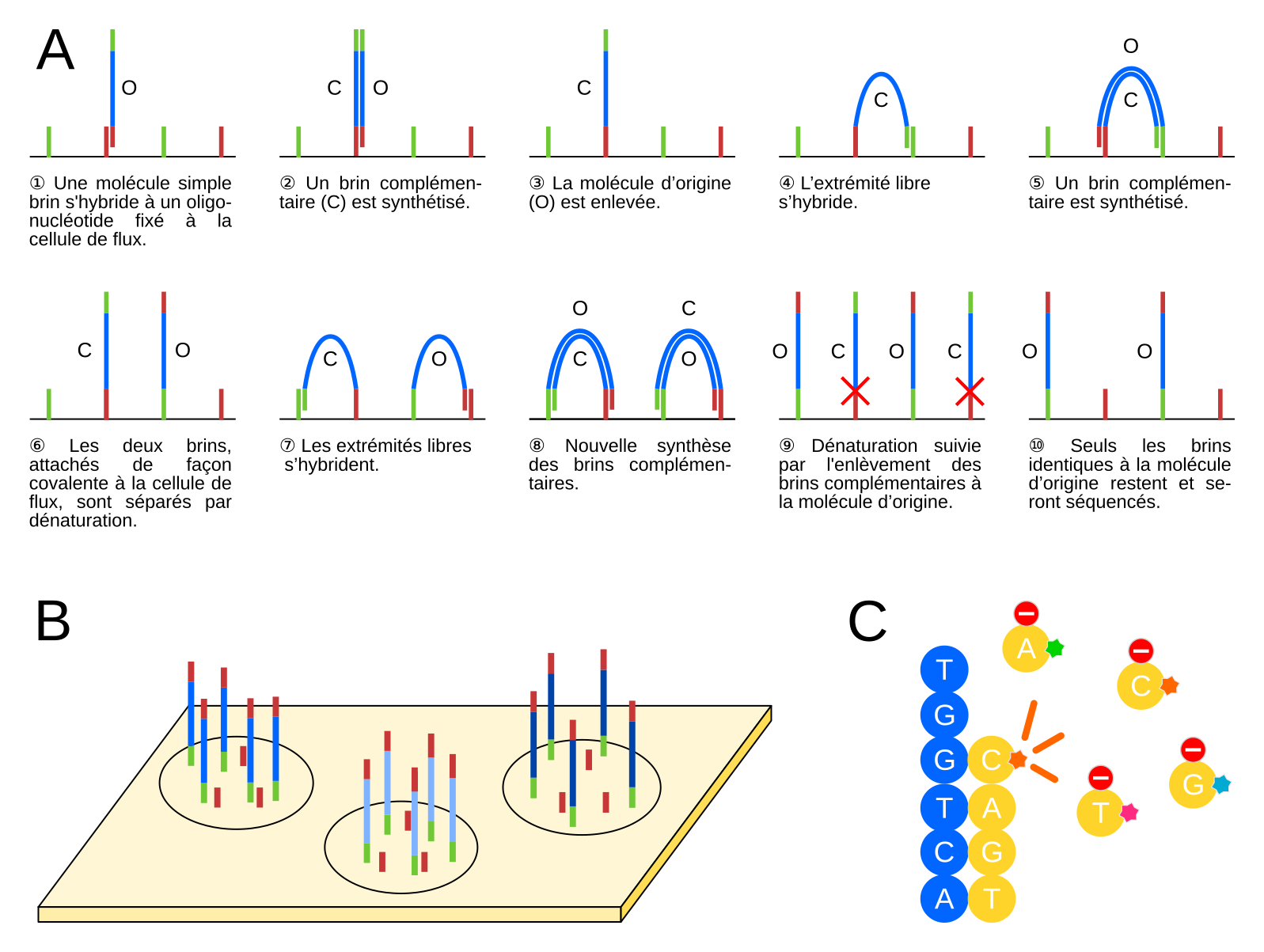
(A) Représentation schématique du processus d’amplification en pont (bridge amplification) des clusters sur la cellule de flux (flow cell, trait noir). Les adaptateurs et les oligonucléotides complémentaires présents sur la cellule de flux sont indiqués en rouge et vert, les fragments d’ADN à séquencer en bleu. La molécule s’étant initalement fixée à la cellule de flux, et les brins dont la séquence est identique, sont notés O ; les brins complémentaires sont notés C. À l’étape 9, les brins complémentaires à la molécule d’origine sont éliminés par clivage chimique d’un nucléotide modifié dans l’oligonucléotide « rouge » fixé à la cellule de flux (croix rouges).
(B) Différentes molécules d’ADN sont amplifiées puis séquencées en même temps sur une même cellule de flux.
(C) Réaction de séquençage. Une polymérase synthétise un brin complémentaire (jaune) au brin situé sur la cellule de flux (bleu) en incorporant des nucléotides qui portent des groupes fluorescents (bleu, rose, orange, ou vert) et un groupe « stop » qui bloque la polymérisation. S’ensuit une étape d’imagerie afin d’identifier le nucléotide incorporé. Ensuite, les groupes fluorescents et « stop » sont enlevés et le processus se répète.
Les applications du NGS
L’apparition des technologies NGS a permis le séquençage des génomes à une vitesse sans précédent et à un coût qui a chuté de façon spectaculaire par rapport à la technologie Sanger (Figure 3). Le Human Genome Project, achevé en 2003 après 13 ans d’effort, a coûté environ 3 milliards de dollars. Seulement six ans plus tard, en 2009, le coût du séquençage d’un génome humain a été réduit à 200 000 dollars et en 2015, la barre symbolique de 1000 dollars a été franchie [4, 5]. Ceci a mis le séquençage des génomes à la portée des laboratoires modestes, et le NGS a ainsi entraîné une révolution dans le domaine de la génomique. En effet, comme illustré dans la Figure 3, le nombre de génomes séquencés a explosé depuis l’arrivée des technologies NGS.
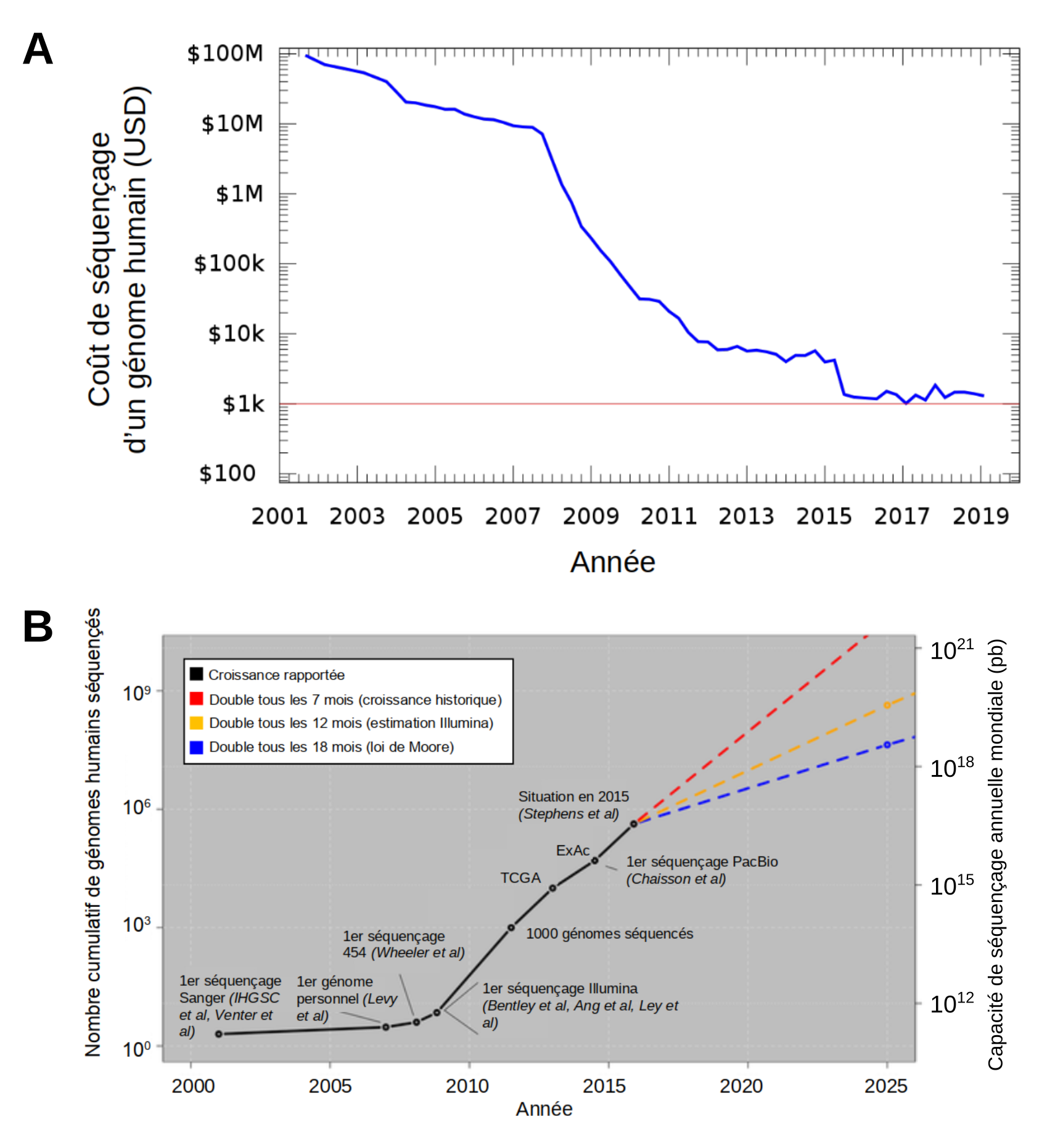
(A) Évolution du coût du séquençage d’un génome humain. Source : Ben Moore, réécrit en gnuplot par grendel|khan, CC0, Wikimedia.
(B) L'explosion du nombre de génomes séquencés depuis l’apparition des technologies NGS. Le nombre total de génomes humains séquencés est indiqué à gauche et la capacité de séquençage annuelle mondiale est à droite. La croissance rapportée d’après les publications scientifiques est illustrée par la ligne noire avec des événements marquants indiqués (grands projets de séquençage TCGA = The Cancer Genome Atlas, ExAC = Exome Aggregation Consortium). Les lignes en pointillés représentent trois extrapolations différentes de la croissance de la capacité de séquençage ; en rouge, la croissance historique, en jaune la croissance estimée par Illumina en 2014, et en bleue la croissance selon la loi de Moore, formulée pour la puissance informatique, qui croît exponentiellement. Source : adapté de Stephens et coll., 2015, CC BY, PLOS Biology.
Les méthodes de séquençage de nouvelle génération, utilisées pour l’étude des génomes, ont été mises à profit pour l’étude des transcriptomes. C’est ainsi que les méthodes de séquençage des ARN (RNA-seq) ont rapidement remplacé les technologies précédentes, qui étaient basées sur des micro-réseaux (microarrays). Ces derniers utilisent des sondes avec des séquences prédéfinies auxquelles les ARN s’hybrident et sont ainsi détectés. Le RNA-seq a plusieurs avantages par rapport à ces micro-réseaux. Premièrement, il permet de détecter et de quantifier des transcrits sans connaissance a priori de leur séquence. Deuxièmement, il autorise la quantification des transcrits sans effet de saturation. En effet, les micro-réseaux étaient limités par le nombre des sondes ; la détection d’un ARN extrêmement abondant pouvait arriver à un plateau suite à une saturation de celles-ci. Avec le RNA-seq au contraire, il n’y a pas de limite de détection quantitative. Troisièmement, le RNA-seq présente moins de bruit de fond que les micro-réseaux. Le NGS est également devenu la technique de référence pour la détection des interactions protéine-ADN par immunoprécipitation de la chromatine (ChIP), car le NGS est plus quantitatif et permet une meilleure résolution que les méthodes qui existaient jusqu’ici.
Dans les années suivantes, le NGS a été utilisé pour des applications de plus en plus diverses. Voici un résumé des applications les plus courantes.
Séquençage des génomes à l’échelle de la population
Bien évidemment, le séquençage des génomes constitue une application majeure du NGS qui a été mentionnée ci-dessus. Mais les technologies NGS ont été développées initialement dans un but encore plus ambitieux : le séquençage d’un très grand nombre de génomes de la population humaine. Les données collectées permettent alors d’identifier les bases génétiques des variations phénotypiques. Ces connaissances sont ensuite utilisées pour des études d’association entre variations génétiques et maladies (genome-wide association studies, GWAS). Ces variations peuvent être de deux types. D’une part, il existe des variations rares qui ont un effet grave sur des caractéristiques simples (par exemple la mucoviscidose, la maladie de Huntington). D’autre part, certaines variations plus fréquentes ont des effets plus légers et pourraient être impliquées dans des phénotypes complexes (par exemple le diabète, les maladies cardiovasculaires). Un des premiers projets était le 1000 Genomes Project, initié en 2008, dans lequel un millier de génomes humains ont été séquencés afin d’établir un catalogue des variantes génétiques ayant une fréquence d’au moins 1 % dans les populations étudiées [6]. Ce projet a été suivi par des initiatives encore plus ambitieuses, par exemple le 100,000 Genomes Project [7].
RNA-seq
Une autre application majeure est le séquençage d’ARN (RNA-sequencing, RNA-seq) qui concerne le séquençage de l’ensemble des transcrits (transcriptome) d’une cellule, d’un tissu ou d’un organisme. Le RNA-seq est majoritairement utilisé pour l’analyse de l’expression différentielle des gènes entre des cellules soumises à des conditions différentes.
Détection des interactions et/ou des modifications des acides nucléiques
Le NGS est aussi un outil très puissant pour la détection d’une grande diversité d’interactions entre moléculrd d'acides nucléiques, entre acides nucléiques et protéines, ainsi que des modifications « épigénétiques » des acides nucléiques1. Une technique couramment utilisée est le « chromatin immunoprecipitation-sequencing » ou « ChIP-seq ». Cette méthode est basée sur la reconnaissance par un anticorps d’un partenaire (souvent une protéine) ou d’une modification épigénétique. Après fragmentation de la chromatine, cet anticorps permet de précipiter spécifiquement les fragments en interaction avec le partenaire d’intérêt, ou qui contiennent la modification épigénétique recherchée. Cela permet de constituer une banque de fragments d’ADN d’intérêt qui sont ensuite séquencés.
Une autre technique très utilisée est celle du « chromosome conformation capture » qui permet l’étude des interactions entre différentes régions du génome. Il existe plusieurs variantes de cette technique, qui permettent soit de détecter des interactions entre deux régions connues (3C), soit des interactions entre une région connue et une ou plusieurs régions inconnues (4C), ou de cartographier toutes les interactions entre des régions a priori inconnues (HiC).
Séquençage ciblé – séquençage des amplicons
Pour un grand nombre d’applications il n’est pas nécessaire de séquencer la totalité du génome ou du transcriptome. Plusieurs techniques ont été développées qui permettent de séquencer uniquement la partie du génome ou du transcriptome qui est la plus intéressante pour répondre à la question posée. Par exemple, une technique couramment utilisée pour étudier des maladies génétiques rares est le séquençage de l’exome (whole exome sequencing, WES), c’est-à-dire de l’ensemble des exons. Sachant que chez l’espèce humaine l’exome constitue uniquement environ 1 % du génome, cette technique permet de réduire énormément la profondeur de séquençage requise et donc de séquencer un très grand nombre d’échantillons avec relativement peu de lectures.
Séquençage des cellules uniques
Le progrès des techniques de séquençage à haut-débit a permis l’apparition des méthodes de séquençage de cellules uniques, avec le RNA-seq comme application majeure. En effet, les méthodes classiques de RNA-seq ne fournissent qu’une moyenne des profils transcriptomiques de toutes les cellules dans une population. À l’inverse, le séquençage ARN sur cellule unique (single cell RNA-seq, scRNA-seq) a pour avantage de montrer la diversité des profils transcriptomiques entre cellules. De plus, le scRNA-seq permet une bien meilleure résolution des profils d’expression des gènes. Par exemple, cette technique peut déterminer si des gènes coexprimés dans une population sont coexprimés dans la même cellule ou dans des cellules différentes.
Une autre possibilité intéressante est le séquençage des génomes de cellules uniques. Ceci permet par exemple d’étudier la diversité génétique entre cellules dans une tumeur et d’étudier son évolution, c’est-à-dire l’accumulation des mutations et réarrangements génétiques au cours de la progression tumorale. Une autre application importante est le séquençage des génomes des microorganismes qui ne peuvent être cultivés en laboratoire.
La troisième révolution : l’apparition des technologies de séquençage long-read
Même si les technologies NGS sont extrêmement puissantes, elles présentent aussi quelques faiblesses, par exemple la faible longueur des fragments séquencés. Des génomes contiennent souvent de nombreuses séquences répétées dont la longueur excède celle des fragments lus par NGS, ce qui mène à des difficultés dans l’assemblage du génome. Par conséquent, un grand nombre de génomes ne sont pas publiés sous la forme d’une séquence continue mais sont disponibles sous une forme très fragmentée, en centaines ou milliers de contigs1. Pour la même raison, les variantes structurales (réarrangements génomiques de plus de 50 paires de bases) entre les génomes de différents individus sont souvent difficiles à détecter. Ceci est un problème important, vu que les variantes structurales sont souvent liées à des maladies génétiques. Un autre problème est que les méthodes NGS ne permettent pas toujours de caractériser des isoformes de transcrits générés par épissage alternatif. De plus, du fait que les méthodes NGS nécessitent une étape d’amplification par PCR, les régions génomiques avec un pourcentage élevé de GC sont souvent sous-représentées, car elles sont moins bien amplifiées par PCR.
Rapidement après l’apparition des méthodes NGS, des technologies dites de troisième génération (TGS, third generation sequencing) ou de lectures longues (long read) sont apparues. Elles se caractérisent par le séquençage, en temps réel2 de molécules uniques [8]. Ces technologies permettent de générer des lectures d’une longueur de plusieurs kilobases voire même des centaines de kilobases. La Figure 4 résume les avantages des technologies long read par rapport au NGS.
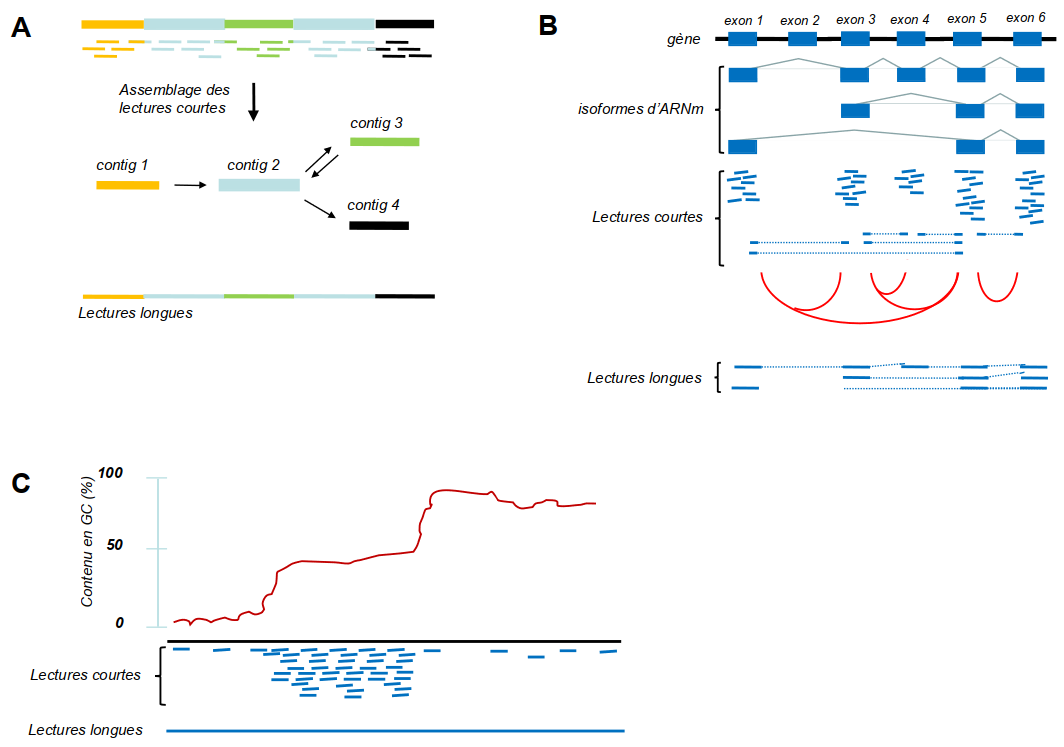
(A) Exemple qui montre le problème des régions répétées (en bleu clair) dans un génome, qui ne peuvent être positionnées à des endroits uniques avec des lectures courtes (NGS). Des lectures qui se situent à l’intérieur de ces régions seront assemblées en un seul contig. La région entre les répétitions (vert) peut être placée en amont ou en aval de ce contig bleu (illustré par les flèches en double-sens), ce qui génère une ambiguïté. Par contre, des lectures longues (long reads, TGS) qui traversent ces régions répétées ne laissent aucune ambiguïté. (B) Plusieurs transcrits (isoformes) peuvent être générés à partir d’un seul gène par épissage alternatif. Des lectures courtes de ces isoformes donneront des lectures à l’intérieur des exons présents dans le mélange (lignes continues) ainsi que des lectures qui chevauchent les jonctions entre les exons (lignes interrompues). Ainsi, les évènements d’épissage alternatif seront détectés ; les jonctions exon-exon détectées dans le mélange sont indiquées par des lignes rouges. Toutefois, les informations concernant les combinaisons des jonctions exon-exon dans les transcrits individuels manquent. Des lectures longues qui couvrent les transcrits entiers fournissent ces informations. (C) Les méthodes NGS dépendent de l’amplification par PCR, ce qui introduit des biais dans des régions pauvres en GC (à gauche de la courbe) ou très riche en GC (à droite). Par conséquent, ces régions seront peu couvertes. Les technologies de troisième génération PacBio et nanopore n’ont pas besoin de l’amplification PCR et présentent donc beaucoup moins de biais avec ce type de régions.
En 2011, Pacific Biosciences lançait sa technologie single molecule real time sequencing (SMRT), basée sur une polymérase attachée au fond d’un micro-puits. Cette polymérase synthétise un brin complémentaire à partir de molécules d’ADN simple brin circulaires (Figure 5). Elle utilise des nucléotides marqués, comme dans la technologie Illumina, mais avec pour différence importante que la synthèse ne s’arrête pas après l’incorporation de chaque nucléotide. La synthèse continue jusqu’au moment où la polymérase s’arrête spontanément (après 10 000 à 100 000 nucléotides incorporés), et l’incorporation des nucléotides est suivie en temps réel dans un film (movie). Un aspect important à mentionner est que cette méthode permet de détecter directement des nucléotides méthylés, car le temps d’incorporation d’un nucléotide modifié est un peu différent de celui d’un nucléotide non modifié. Le taux d’erreurs de cette méthode est assez élevé pour une seule lecture (environ 10 %), mais du fait de la circularité de la molécule d'ADN à séquencer, la polymérase fait le tour plusieurs fois, ce qui augmente énormément la fiabilité jusqu’à des valeurs comparables ou même supérieures à celles de la technologie Illumina.
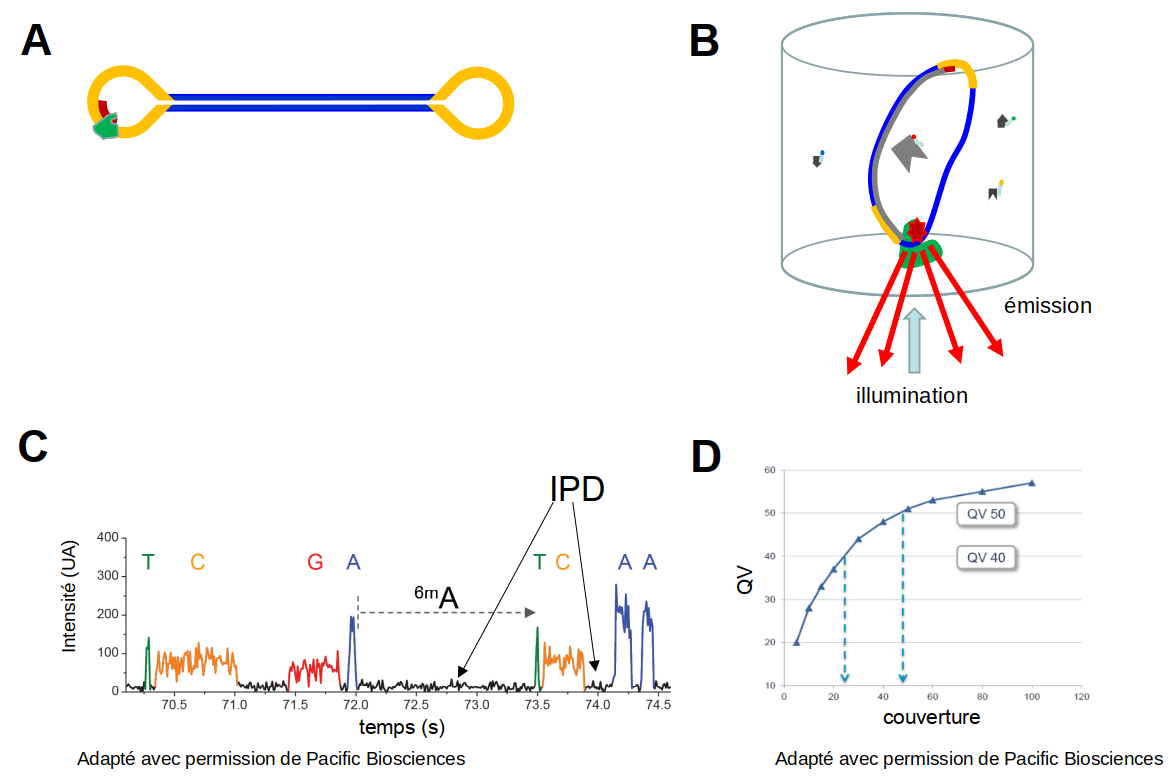
(A) Pendant la préparation de la banque, des adaptateurs en tige boucle (jaune) sont attachés à des molécules d’ADN double-brin (bleu), ce qui donne des molécules circulaires (SMRT bells). Ensuite, une amorce (rouge) et une polymérase (vert) se fixent sur l’adaptateur. (B) Représentation schématique d’un puits dans lequel le séquençage a lieu ; la polymérase, qui se fixe sur le fond du puits, incorporera des nucléotides fluorescents, et à chaque évènement d’incorporation un signal fluorescent (« émission ») sera généré après illumination par un laser. Ces signaux sont enregistrés par une caméra en temps réel, ce qui génère un film (movie). (C) Exemple de movie. Les différents nucléotides sont identifiés par leur couleur ; le temps entre les incorporations successives est enregistré également (interpulse duration – IPD, en noir). On remarque que dans cette technologie de séquençage, la vitesse de polymérisation (2 à 4 nucléotides par seconde) est bien plus faible qu’in vivo. La présence d’une modification épigénétique, comme la 6-méthyladénosine (6mA) est responsable d'un IPD plus long, ce qui permet son identification. (D) La qualité des données de séquençage (quality value – QV) augmente fortement quand la polymérase fait plusieurs fois le tour des molécules circulaires. Par exemple, quand la polymérase fait 25 fois le tour, la précision atteint 99,999 % (QV40) et pour 50 tours, la précision atteint 99,9999 % (QV50), ce qui correspond à un taux d’erreur inférieur à celui du séquençage Illumina.
En 2014, la méthode d’Oxford Nanopore Technologies (ONT) est apparue sur le marché. La technologie nanopore est unique car elle n’utilise pas de polymérase pour la synthèse d’une copie de la molécule à séquencer. La séquence d’une molécule d’ADN ou d’ARN est directement déduite grâce à la perturbation d’un courant électrique traversant un « nanopore » incorporé dans une membrane qui sépare deux compartiments contenant des solutions ioniques (Figure 6). La perturbation du signal est une fonction de la composition des nucléotides présents dans le pore à chaque instant, et c’est en suivant en temps réel les successions de ces perturbations qu’on peut en déduire la séquence. Comme pour la technologie SMRT de Pacific Biosciences, on peut détecter directement les modifications des nucléotides car celles-ci peuvent perturber le signal électrique de façon spécifique.
La technologie nanopore peut générer des lectures extrêmement longues, qui peuvent aller jusqu’à 1 Mb ou même plus. A priori le seul facteur limitant serait la longueur des fragments d’ADN que l’on réussit à obtenir lors de l’extraction de cette molécule. En effet, les molécules d’ADN génomique sont généralement fragmentées pendant cette étape. Un vrai défi est donc de trouver des méthodes d’extraction qui laissent l’ADN le plus intact possible.
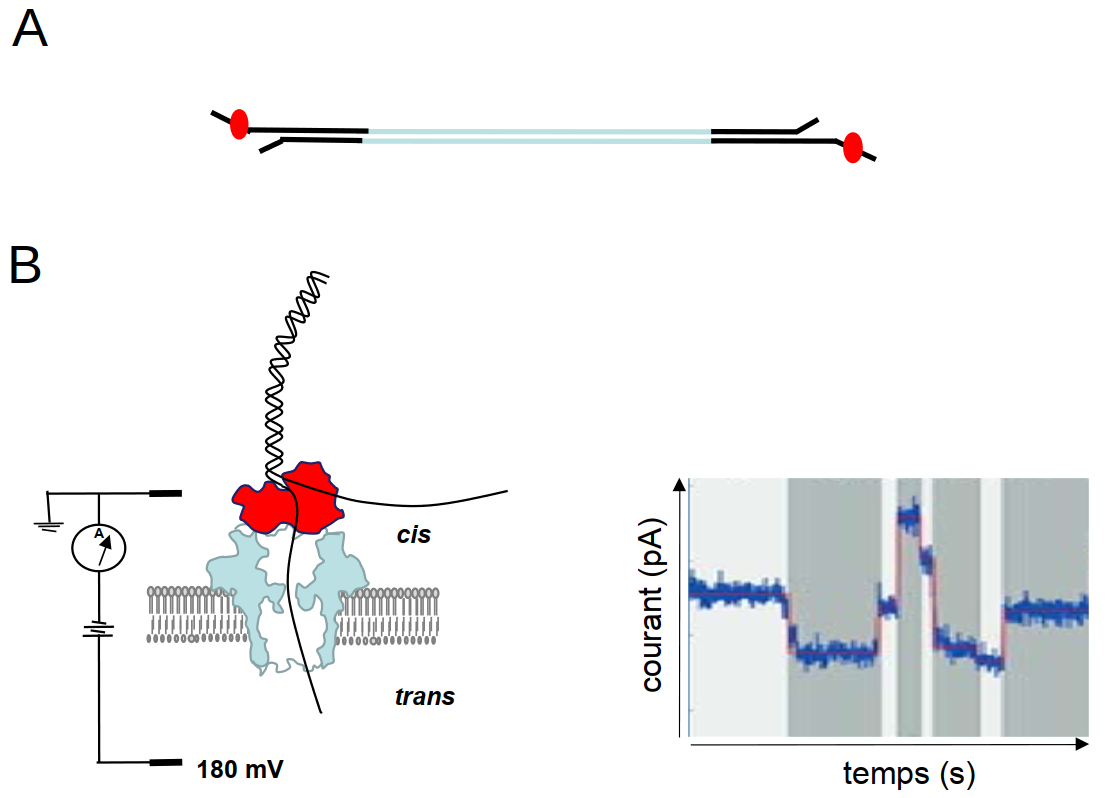
(A) Dans une banque nanopore, des adaptateurs (en noir) sont attachés aux molécules d’ADN double-brin à séquencer (en bleu), comme pour les banques Illumina. Sur chaque adaptateur, une protéine motrice (rouge) est fixée. (B) Dans une cellule de flux d’Oxford Nanopore Technologies (ONT), deux compartiments (cis et trans) contenant des solutions ioniques sont séparés par une membrane qui contient des pores ménagés par des protéines. Un acide nucléique (noir) traverse le pore (en bleu) de façon contrôlée grâce à la présence de la protéine motrice (rouge). Avant de traverser le pore, l’acide nucléique est dénaturé par la protéine motrice et ainsi un seul des deux brins traverse le pore. Quand une molécule d’ADN ou d’ARN traverse le pore, le courant électrique est modifié ; celui-ci est enregistré en temps réel et graphiquement représenté par un squiggle plot (à droite).
Traitement des données de séquençage : le rôle de la bio-informatique.
Les technologies NGS et TGS ont nécessité le développement d’outils de bio-informatique spécifiques, capables d’analyser les données générées par les différentes méthodes. Un grand nombre d’algorithmes capables de gérer des lectures courtes NGS ainsi que des algorithmes permettant l’assemblage de génomes de novo, la détection des polymorphismes mononucléotidiques (SNP), l’analyse ChIP-seq et RNA-seq ont été développés [9, 10]. Des programmes corrigeant les biais introduits pendant la préparation des banques ont également été mis au point. L’ensemble des améliorations expérimentales et algorithmiques ont fortement renforcé l’utilité des technologies NGS. L’apparition des technologies de troisième génération a également nécessité le développement d’outils adaptés permettant de gérer des lectures (très) longues et des taux d’erreurs élevés [11].
Conclusions et perspectives
Il y a un peu plus de quarante ans, l’apparition de la technologie de séquençage Sanger a permis pour la première fois de déchiffrer des gènes et des génomes, ce qui a révolutionné le domaine de la génomique et de la biologie en général. Dans les années 2000, le développement des méthodes NGS, beaucoup plus puissantes que le séquençage Sanger, a fait chuter les prix du séquençage de façon considérable et a mis le séquençage des génomes à la portée des laboratoires modestes. Plus récemment encore, le développement et la mise au point des technologies de troisième génération, ou long read, a ouvert des possibilités nouvelles permettant le séquençage des génomes avec une qualité sans précédent.
Nous disposons donc aujourd’hui d’une boite à outils extrêmement puissante pour l’exploration des génomes et les années récentes ont vu de nombreuses découvertes excitantes qui vont sans doute continuer dans les années à venir.
Malgré les progrès impressionnants accomplis ces dernières années, il reste des défis majeurs pour l’avenir. Au niveau de l’analyse bio-informatique des quantités massives de données de séquençage, un objectif important sera de développer des outils plus performants et rapides, notamment dans le cadre des utilisations cliniques, où chaque jour compte. Il y a aussi des questions importantes concernant la gestion et l’utilisation des données de séquençage : comment stocker et protéger ces données, quelles informations faut-il partager et comment, etc.
Afin d’obtenir une compréhension de la cellule plus profonde et complète, un défi majeur pour l’avenir sera de réussir à mettre en relation les résultats de la génomique avec les données épigénomiques, transcriptomiques et protéomiques. Alors qu’il existe aujourd’hui des outils puissants et rapides pour l’étude des génomes, épigénomes et transcriptomes, l’exploration des protéomes reste techniquement très délicate. Il est intéressant dans ce cadre de mentionner que la technologie nanopore permettrait, a priori, de séquencer des protéines [12], mais à ce jour cette application n’est pas encore opérationnelle. La mise au point d’une méthode rapide et économique pour séquencer l’ensemble des protéines d’une cellule, tissu ou organisme marquera sans doute la prochaine révolution dans l’« omique ».
Références
[1] About the Human Genome Project, Human Genome Project Information Archive
[2] Elaine R. Mardis. The impact of next-generation sequencing technology on geneticsTrends in Genetics, 2008, vol 24, n° 3 https://doi.org/10.1016/j.tig.2007.12.007
[3] Sara Goodwin, John D. McPherson and W. Richard McCombie. Coming of age: ten years of nextgeneration sequencing technologies, Nat. Rev. Genet. 2016, 17:333-351
[4] The $1,000 Genome, infographie d'Illumina
[5] Genome Sequencing Stocks On The Rise, Ken Berman, Forbes, 21 février 2019
[6] The International Genome Sample Resource
[7] The 100,000 Genomes Project, Genomics england
[8] Erwin L. van Dijk, Yan Jaszczyszyn, Delphine Naquin, and Claude Thermes. The Third Revolution in Sequencing Technology. Trends in Genetics, 2018, vol. 34, n° 9 https://doi.org/10.1016/j.tig.2018.05.008
[9] Hatem, A. et al. Benchmarking short sequence mapping tools. BMC Bioinformatics, 2013, 14, 184
[10] Rucha M. Wadapurkar, Renu Vyas. Computational analysis of next generation sequencing data and its applications in clinical oncology (2018). https://doi.org/10.1016/j.imu.2018.05.003
[11] Shanika L. Amarasingh, Shian Su, Xueyi Dong, Luke Zappia, Matthew E. Ritchie and Quentin Gouil. Opportunities and challenges in long-read sequencing data analysis. Genome Biology, 2020, 21:30 https://doi.org/10.1186/s13059-020-1935-5
[12] Kolmogorov M, Kennedy E, Dong Z, Timp G, Pevzner PA. Single-molecule protein identification by sub-nanopore sensors. PLoS Comput Biol. 2017;13(5):e1005356. doi:10.1371/journal.pcbi.1005356




{kind=link}
{kind=link}
{kind=link}