

Les organismes vivants les mieux adaptés à survivre et se reproduire sont les grands gagnants du processus de sélection naturelle. Les maladies infectieuses exercent une forte pression de sélection sur le système immunitaire, contribuant à éliminer des allèles délétères et à sélectionner positivement les variants amenant un avantage en termes de survie. Du fait que l’humanité a payé un tribut énorme à la peste en nombre de morts durant les pandémies historiques, la bactérie Yersinia pestis, hautement pathogène, a probablement favorisé la sélection de tels variants génétiques chez les survivants. Les méthodes pour analyser ces effets ont considérablement évolué durant les dernières décennies avec l’arrivée du séquençage de génome à haut débit, et l’explosion de la puissance de calcul des ordinateurs, amenant les approches de génétique des populations telles que les études d’association pangénomiques à prendre le pas sur les approches ciblées sur des gènes candidats déterminés a priori. Dans cette revue, les études anciennes et récentes et les hypothèses sur la sélection naturelle par la peste sont présentées et les mécanismes immunitaires correspondants sont décrits lorsqu’ils sont connus. Une série d’allèles qui auraient été sélectionnés par la peste sont associés à une sensibilité accrue à d’autres maladies, mettant l’accent sur l’impact des mutations affectant les gènes liés à l’immunité. Finalement, les effets de la sélection naturelle sur le pathogène lui-même et les animaux réservoirs sont présentés.
Dans son ouvrage L’origine des espèces, Charles Darwin a proposé en 1859 que les caractéristiques des êtres vivants évoluaient de génération en génération, et que la principale force motrice de cette évolution était la sélection naturelle exercée par l’environnement. Selon ce principe, les sujets les plus adaptés survivent et se reproduisent, alors que les sujets les moins adaptés sont désavantagés (1). En conséquence, les caractéristiques (le phénotype) des plus adaptés deviennent plus fréquentes dans la population. Les variations génétiques spontanées et héritables (mutations ponctuelles et réarrangements de l’ADN) expliquent l’apparition de nouveaux traits, en même temps que les caractéristiques de l’environnement telles que les ressources en eau et nourriture, la température et l’hygrométrie, ou les infections, déterminent qui est le plus adapté.
Les chances de survivre aux infections, ou simplement d’assurer sa descendance, diffèrent considérablement selon qu’un individu a un allèle avantageux ou délétère d’un gène permettant de résister à une maladie. Les micro-organismes pathogènes représentent l’une des plus fortes sources de pression de sélection sur le génome humain (2, 3) et génèrent probablement la plus grande part de la variabilité observée chez les individus d’aujourd’hui face à l’infection (4). La peste et les autres maladies infectieuses mortelles (malaria, tuberculose, choléra, variole, etc.) ont toutes été soupçonnées d’avoir exercé une sélection positive ou négative sur le génome humain (3). La sélection est dite négative lorsqu’elle élimine des allèles délétères d’une population, et positive lorsqu’elle fait ressortir et favorise des allèles avantageux.
Les barrières géographiques ont toujours contribué à isoler les populations, créant des environnements distincts dans lesquels sévissent des pathogènes différents. Dans chaque environnement, les populations s’adaptent aux agents pathogènes qui les menacent, tout en restant sensibles aux infections absentes mais existant ailleurs. Ainsi, la sélection naturelle contribue à la variation géographique (5). L’introduction de maladies nouvelles, telle que la peste venue d’Asie et ayant ravagé l’Europe durant deux grandes pandémies, contribue à façonner le génome des populations affectées. À l’aide des récits de ces épisodes de peste, il est possible d’identifier les signatures de sélection en comparant les génomes et phénotypes de populations ayant des historiques d’infection différents.
La survie aux infections dépend essentiellement (mais pas seulement) de la capacité du système immunitaire à affronter et tuer les microbes l’agressant. Ainsi, la sélection naturelle ayant agi autrefois sur les ancêtres des individus contemporains génère chez ces derniers des différences dans l’expression de gènes et de réponses immunitaires face aux agents infectieux, qui font que des individus issus de régions distinctes de la planète diffèrent dans leur sensibilité aux infections (6). Sans surprise, les allèles dont la sélection positive par les infections a pu être mise en évidence avec le plus de certitude ont principalement été des gènes de l’immunité, impliqués dans les défenses de l’hôte contre les infections (7).
Selon l’idée que les infections ont sélectionné positivement ou négativement des allèles liés à l’immunité, les épidémies et pandémies devraient générer des populations exemptes des allèles délétères, avec de ce fait un meilleur système immunitaire et une espérance de vie plus longue. Pourtant et de manière surprenante, l’adaptation à une infection particulière n’implique pas une adaptation globale, et des traits génétiques entraînant la sensibilité à une maladie peuvent être favorisés s’ils confèrent un avantage face à une autre pression de sélection. Par exemple, la malaria (ou paludisme) a favorisé chez les humains exposés plusieurs défauts érythrocytaires d’origine génétique parce qu’ils protègent contre le parasite à l’origine de la maladie (8).
Les premières études des conséquences des maladies infectieuses sur la génétique humaine datent de plusieurs décennies et remontent à une époque où les chercheurs n’avaient pas accès à la séquence complète du génome humain. L’idée que les infections ont un pouvoir de sélection sur le génome date des années 1950, lorsqu’il a été observé que l’anémie falciforme (drépanocytose) avait un effet protecteur contre les formes graves de malaria causée par Plasmodium falciparum dans des populations africaines, et que l’incidence de l’anémie falciforme et d’autres défauts des globules rouges coïncidait avec la distribution géographique de la malaria, suggérant que ces anomalies génétiques pourraient conférer une certaine résistance à la maladie (4). Par conséquent, les premières études suivaient une hypothèse basée sur des observations phénotypiques, et utilisaient des outils d’analyse et des statistiques simples. La compréhension des mécanismes biologiques à l’origine de la résistance était parfois absente ou se limitait à des hypothèses. Avec les progrès de la biologie moléculaire et de la génomique, des paramètres plus précis ont pu être évalués dans le cas de la peste, tels que des mutations connues pour leurs conséquences cliniques. On verra ci-dessous les exemples des gènes CCR5, ou FPR1, associés à des maladies.
Plus récemment, l’avènement de méthodes de séquençage d’ADN rapides et à haut débit (next-generation sequencing) et l’explosion des capacités de calcul des ordinateurs ont rendu possible l’analyse de génomes entiers. Elles ont permis le développement de la génétique des populations, dont le but est de comprendre les processus évolutifs qui ont façonné la diversité des populations contemporaines (7). Dans les études d’association pangénomiques (genome-wide association studies, ou GWAS), des génomes entiers peuvent maintenant être examinés sans a priori sur un gène ou un mécanisme. Cette augmentation de la puissance d’analyse s’est traduite par une possibilité améliorée d’identifier les causes et parfois de réévaluer des conclusions précédentes. Les sections successives de la présente revue reflètent cette évolution des méthodes, et les auteurs ont essayé de présenter une description objective des études disponibles.
La peste, un fléau historique à l’origine de pandémies
Yersinia pestis, l’agent de la peste, représente l’un des pathogènes les plus dévastateurs de l’histoire de l’humanité. Les techniques récentes d’analyse de l’ADN ancien ont permis de montrer que Y. pestis a émergé en Asie il y a 5 700 à 6 000 ans où elle infectait déjà les Hommes préhistoriques des âges de la pierre et du Bronze (9-11). Ensuite, Y. pestis s’est propagé à travers l’Europe, l’Asie, l’Afrique centrale et du Nord durant deux pandémies majeures. En effet, bien qu’il soit surtout connu pour la mort noire du Moyen Âge, Y. pestis a d’abord été à l’origine d’une première pandémie ayant affecté le pourtour méditerranéen. Débutée au cours du VIe siècle et ayant eu des répliques jusqu’au VIIIe siècle, la peste de Justinien ravagea l’Empire romain d’Orient et en accéléra la fin. Plus tard, la mort noire, ou peste noire, a envahi l’Europe de 1347 à 1351, et aurait mis un coup de frein brutal au rayonnement économique et culturel du continent. Cette pandémie a connu de multiples vagues jusqu’au XVIIIe siècle, avant de quitter la région. L’analyse du génome des victimes les plus récentes a suggéré que des foyers auraient persisté localement durant au moins trois siècles, et auraient causé ces multiples résurgences (12). La diffusion de la maladie a suivi les routes de l’activité humaine (13). Avant la révolution industrielle, les déplacements à longue distance par voie de terre, de rivière, ou maritimes servaient principalement au commerce. En conséquence, la peste est toujours présente le long de chemins traditionnels du négoce tels que la route de la soie. Les voies maritimes, elles, ont amené la peste par le sud de l’Asie jusqu’au Moyen-Orient, et de là en Europe. Ces deux pandémies auraient ensemble éliminé 200 millions d’Européens, et jusqu’à 40 % de la population au Moyen Âge (13).
À l’ère moderne, la maladie s’est encore répandue dans le monde entier lors d’une troisième pandémie plus globale mais de moindre mortalité, démarrée à Hong-Kong en 1894. Par les moyens modernes de transport, la peste a voyagé à travers le monde et a atteint des endroits auparavant épargnés tels que les Amériques ou Madagascar. Elle est entrée à San Francisco par bateau en 1900 et s’est installée dans des États de l’Ouest des États-Unis où des cas humains ont occasionnellement lieu, pour un total heureusement limité à un peu plus de mille cas sur l’ensemble du pays (14). Elle est aussi entrée à l’époque en Amérique du Sud, mais aucun cas n’y est plus enregistré depuis des décennies. En plus des cas rares aux États-Unis, la peste est active dans des foyers d’Afrique et d’Asie où de récentes flambées ont eu lieu. Ces foyers actuellement actifs sont considérés comme des signes que la troisième pandémie est toujours en cours. À cause de cas récents dans des pays longtemps épargnés, l’Organisation mondiale de la santé et les centres pour le contrôle et la prévention des maladies (CDC) considèrent maintenant la peste comme ré-émergente (15). C’est au cours de la dernière pandémie qu’Alexandre Yersin a découvert le bacille agent de la maladie et qui porte son nom, Yersinia pestis.
Des loci présumés liés à la peste, finalement mis hors de cause
La compréhension du fait que les maladies infectieuses constituent des pressions de sélection a entraîné l’idée que la peste avait certainement contribué à l’évolution par sélection naturelle des populations humaines, et a donc été la coupable idéale d’une série d’hypothèses. Toutefois, il faut prendre garde ne pas tirer de conclusions hâtives. En effet, mis à part les épisodes frappants tels que la peste noire, d’autres pressions de sélection existent, parmi lesquelles les autres maladies mortelles, les problèmes de fertilité, ainsi que les infections moins mortelles mais exerçant une pression permanente sur les populations durant de plus longues périodes de temps. Ainsi, la sélection réelle d’allèles par ces facteurs a pu être attribuée, à tort, à la peste. Des preuves expérimentales sont nécessaires pour conclure, et cela n’a pas toujours été le cas.
Cependant il est très probable que la peste ait réellement modifié la variabilité génétique humaine : elle a tué des humains par dizaines de millions, et cela quel que soit leur âge, faisant que nombre d’entre eux n’ont pas eu de descendance (16). La sélection a probablement éliminé certains allèles conférant une sensibilité à la maladie et favorisé les allèles permettant une résistance. Illustrant ce point, une analyse des cimetières de Londres a révélé que les gens ayant vécu après la peste noire ont bénéficié d’une espérance de vie supérieure à celle des personnes ayant vécu durant la période précédant l’épidémie, et cela malgré des résurgences répétées de peste dans les siècles suivants (17). Enfin, du fait que la peste a affecté certaines parties du globe et pas d’autres, la recherche de caractères sélectionnés a souvent consisté en une comparaison de populations touchées ou non durant leur histoire.
Les groupes sanguins
Les groupes sanguins ont été parmi les premières caractéristiques moléculaires de sous-populations humaines mises en évidence par la médecine moderne. Les groupes A, B, AB et O correspondent à la présence des antigènes oligosaccharidiques A ou B ou les deux sur les globules rouges ; le groupe O n’ayant ni A ni B, mais seulement leur précurseur moléculaire H. La présence ou l’absence du facteur Rhésus ou du facteur Lewis s’ajoutent à ces différences. Chaque sujet produit des anticorps contre les antigènes qu’il n’a pas, et lorsque des microbes portent des antigènes ressemblant aux marqueurs sanguins, les anticorps contribuent à la réponse de l’hôte. Par ailleurs, de nombreux microbes pathogènes (bactéries et virus) interagissent avec les marqueurs sanguins, et cela peut contribuer à la virulence ou au contraire faciliter l’immunité de l’hôte (voir la revue complète de Cooling (18)).
Ces groupes ne sont pas répartis de manière homogène dans la population mondiale. Bien que les chiffres varient selon les sources, le groupe le plus fréquent est O+, qui représente 38 % de la population mondiale, suivi par le groupe A+ présent à une fréquence comprise entre 27 et 34 %. Les autres groupes sont présents à moins de 10 %, le plus rare étant AB– (1 %) alors que les Inuits et les Amérindiens sont presque exclusivement O (19) et que les Asiatiques ont la plus forte proportion de B. L’observation de telles disparités géographiques a fait penser qu’elles pourraient être les conséquences des épidémies du passé.
Bien que le type O soit le plus fréquent au monde, il est moins fréquent en Europe, au Moyen-Orient et en Asie, les zones géographiques les plus affectées par les deux grandes pandémies de peste, alors qu’il est plus abondant en Amérique du Sud, le continent le moins touché par la peste (19). Dans les années 1950-60, la peste a été considérée comme ayant sélectionné négativement le groupe O, car une forte réduction du groupe O avait été observée dans des squelettes anglais du XIVe siècle datant d’après la peste noire, par rapport à ceux d’avant. Cependant, aucune réactivité particulière de Y. pestis avec les antigènes ABH ou Lewis n’a pu être mise en évidence in vitro (18). Une controverse persiste concernant la présence sur le bacille d’un antigène de type H, qui pourrait avoir mené à une sélection (20). En conclusion, des effets de la peste sur les groupes sanguins ont été évoqués sur la base de leur fréquence chez les populations exposées ou non à la peste, mais cela n’a pas été confirmé par l’identification d’un mécanisme microbiologique ou par la génétique des populations.
L’hémochromatose : sélection positive ou négative ?
Le fer est essentiel à la multiplication de nombreuses bactéries pathogènes. Chez un sujet sain, le fer libre est maintenu à un taux très faible par des protéines le capturant, par exemple la transferrine (Tf) et la lactoferrine. Chez la souris, l’augmentation expérimentale de la quantité de ces deux protéines permet de contrôler une infection pesteuse à faible dose (21).
L’hémochromatose héréditaire est une maladie due à une accumulation progressive de fer dans les organes, ce qui facilite la multiplication bactérienne et donc les infections, ce qui peut mener à terme à la mort des sujets atteints d’hémochromatose héréditaire. En 2011, un chercheur de l’université de Chicago est mort d’infection par une souche de Y. pestis pourtant atténuée par la perte d’un système de capture de fer nommé yersiniabactine. Ce système, parmi les plus efficaces connus, est capable d’enlever le fer des protéines de l’hôte (22). L’autopsie a révélé que ce chercheur souffrait d’hémochromatose héréditaire, et que son haut taux de fer avait permis à la bactérie de trouver le fer nécessaire pour restaurer une virulence complète (23, 24).
Toutefois et de manière surprenante, l’hémochromatose héréditaire est la maladie portant sur un seul gène la plus courante chez les populations originaires d’Europe du Nord et de l’Ouest, et 80 % des personnes atteintes sont homozygotes pour un allèle HFE muté (codant HFE, une protéine régulant l’absorption du fer). Cette mutation est absente chez les Africains, Asiatiques et Australasiens. Moalem et ses collaborateurs ont proposé que les épidémies de peste en Europe ont pu sélectionner positivement cette mutation (25). L’hypothèse est basée sur le fait que HFE s’associe au récepteur de la transferrine, ce qui fait que chez les patients atteints d’hémochromatose héréditaire, l’absence d’HFE à la surface des cellules cause des taux bas de fer dans les macrophages, un état qui empêcherait la multiplication intracellulaire de Y. pestis et donc réduirait le risque d’infection systémique fatale. Ce bénéfice ne s’appliquerait que chez les sujets jeunes, avant que le fer ne s’accumule dans les tissus. Toutefois, pour valider cette hypothèse, il faudrait démontrer qu’un faible taux de fer dans les macrophages suffit à prévenir la multiplication d’une Y. pestis non atténuée, car le système yersiniabactine est très efficace pour capturer le fer.
La mutation CCR5-∆32 et la résistance au VIH
L’infection mondiale par le virus de l’immunodéficience humaine (VIH) a amené de nombreuses recherches sur sa dissémination. Il a été rapporté à l’époque que, de manière surprenante, certaines personnes restaient séronégatives malgré une exposition fréquente au virus. La raison a été trouvée au niveau génétique : elles présentaient une mutation du gène codant la protéine CCR5, un récepteur de signaux chimiotactiques. CCR5 a alors été identifié comme une des molécules utilisées par le virus pour entrer dans les cellules, avec l’antigène CD4 présent sur les lymphocytes T (26). Chez les personnes hétérozygotes pour la mutation CCR5-∆32, l’infection était ralentie, et les personnes homozygotes étaient protégées. Par ailleurs, les chimiokines se liant à CCR5 (RANTES et MIP-1α/ß) inhibent l’infection des cellules par le virus. Les médias ont aussi beaucoup commenté le fait que deux malades du sida avaient connu une rémission ou une guérison après avoir reçu une greffe de moelle osseuse venant d’un donneur porteur de la mutation CCR5-∆32 (connus dans les médias sous les noms de « patient de Berlin » et « patient de Londres »).
Un résultat surprenant a été que cette mutation CCR5-∆32 est présente à une fréquence plus élevée (10 %) dans la population d’Europe du Nord (de « type caucasien »), alors qu’elle est quasi-absente en Asie, Afrique, Moyen-Orient et chez les Amérindiens (27). Cette répartition a alors été interprétée comme le résultat d’une sélection positive sur certaines populations (28, 29). Une telle sélection impliquait que CCR5-∆32 constitue un avantage face à un facteur de sélection causant une mortalité élevée. L’hypothèse a alors été émise que la mutation CCR5-∆32 pourrait aussi protéger contre la peste.
Cette hypothèse a été testée en reproduisant la mutation chez la souris. Plusieurs travaux ont alors montré que les souris ∆CCR5 (chez qui le CCR5 a été enlevé par génie génétique) n’étaient pas protégées contre l’infection par Y. pestis, même si leurs macrophages étaient moins infectés (30). La mutation CCR5-∆32 ne protégeant pas contre la peste (en tout cas chez la souris), cette maladie ne doit pas être à l’origine de la sélection de cette mutation. En 2003, Galvani et ses collaborateurs ont rapporté que la variole serait un meilleur candidat pour la sélection de CCR5-∆32 (31). Finalement, cette mutation serait nettement plus ancienne que la grande peste noire, puisqu’elle a été observée chez des squelettes datant de l’âge du Bronze (32).
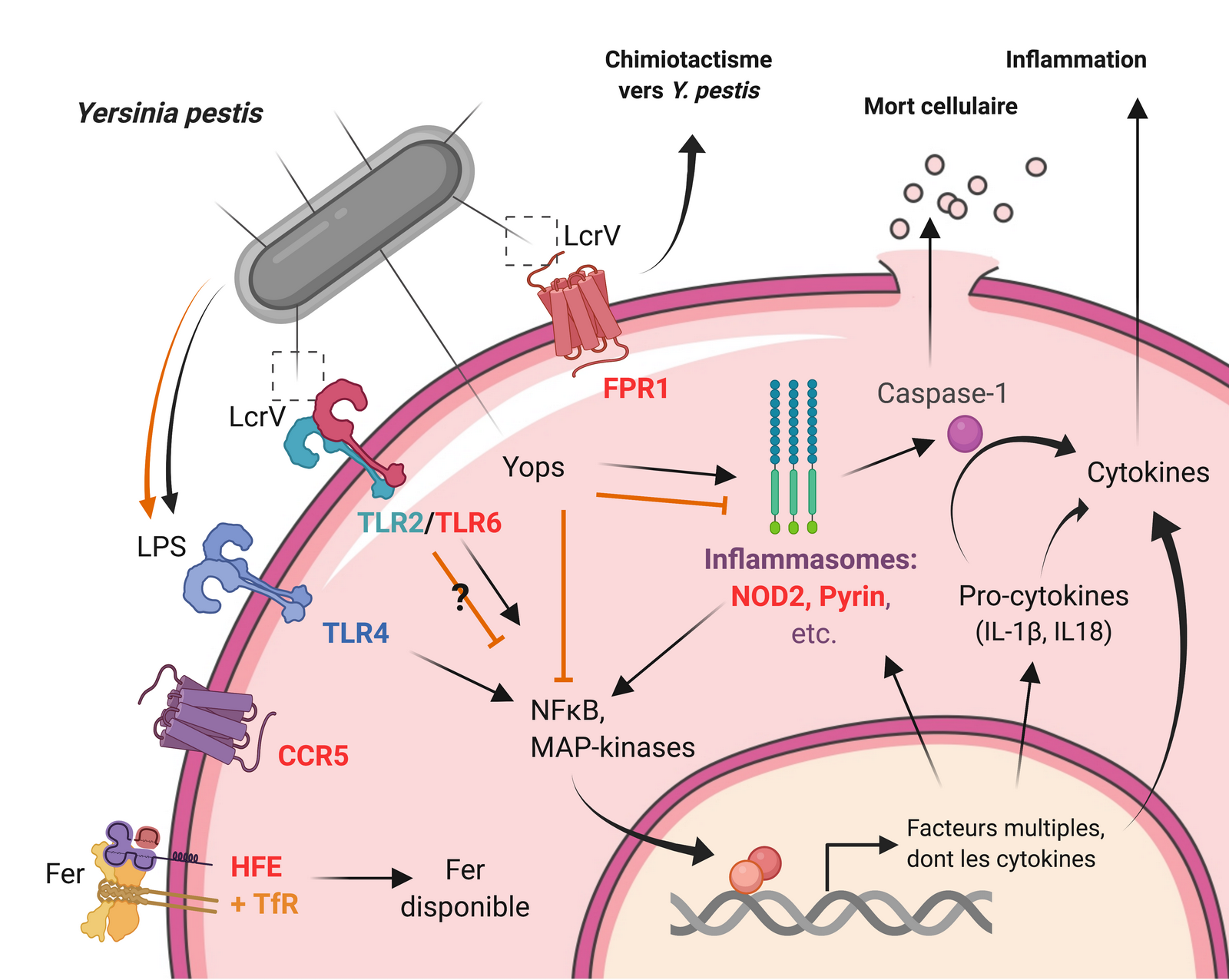
Représentation schématique des voies de signalisation intracellulaires activées après détection de Y. pestis par un macrophage (flèches noires pleines) et les mécanismes de virulence utilisés par Y. pestis pour s’y opposer (lignes orange), avec les facteurs de l’hôte discutés dans le texte (noms en rouge). Les aiguilles du système de sécrétion de type III sont représentées par des traits noirs partant de la membrane plasmique de la bactérie. L’interaction du LPS avec TLR4 est notée activatrice et inhibitrice parce que Y. pestis modifie l’acylation de son LPS à 37 °C, de hexa-acylé (activateur) à tétra-acylé (antagoniste) du fait de la mutation LpxL. De même les signaux donnés par LcrV via TLR2/6 restent à préciser. Une notation simplifiée identique a été utilisée pour indiquer que l’action des Yops sur les inflammasomes est principalement inhibitrice, bien que l’inhibition des Rho GTPases par YopT et YopE active la pyrine.
Le gène codant la caspase 12
Les caspases forment une famille de protéines intracellulaires impliquées dans des processus de mort cellulaire programmée et d’inflammation. Le gène codant la caspase 12 existe principalement chez l’Homme sous une forme tronquée Csp12S, alors que les populations d’origine africaine ont principalement la forme active Csp12L. Cette caspase a une activité anti-inflammatoire qui diminue la capacité des cellules à répondre au lipopolysaccharide (LPS) bactérien, et de ce fait le gène non tronqué est associé à un risque plus élevé de sepsis, le choc inflammatoire d’origine infectieuse. Le gène tronqué a donc probablement bénéficié d’une pression de sélection positive importante et un rôle de la peste a été évoqué (33). Pour tester cette hypothèse, la contribution de la caspase 12 a été évaluée in vitro dans le modèle de réponse des macrophages, cellules centrales de l’immunité innée contre Y. pestis, à l’aide de cellules où le gène csp12 était inactivé. Toutefois, la conclusion a été que la caspase 12 ne participe pas à la réponse cellulaire anti-pesteuse (34), ce qui rend peu probable un rôle dans la sélection de Csp12L.
Sélection génétique par la peste et relation avec d'autres pathologies
En une décennie, des centaines de loci associés à des maladies chroniques inflammatoires ou auto-immunitaires ont été identifiés (6). Nombre de ces gènes sont aussi impliqués dans la sensibilité aux maladies infectieuses, amenant à penser qu’un réseau restreint de gènes intervient dans la réponse à de multiples maladies. Des loci communs ont ainsi été trouvés entre plusieurs maladies telles que la maladie cœliaque, la sclérose en plaques, l’arthrite rhumatoïde, la maladie de Crohn, le psoriasis et le lupus (35). De plus, les loci affectés sont souvent situés à proximité de gènes impliqués dans le fonctionnement du système immunitaire, amenant à penser que la sélection de tels loci pourrait être le fait de facteurs de sélection mobilisant le système immunitaire, notamment les infections. Ainsi, les maladies chroniques inflammatoires ou auto-immunitaires pourraient être une conséquence négative d’une protection accrue contre les infections. Certaines mutations, bien étudiées pour leur rôle dans d’autres pathologies, confèrent une protection contre la peste et il a été proposé qu’elles aient été sélectionnées par cette maladie.
FPR1, un « récepteur de la peste »
La bactérie Y. pestis doit en grande partie sa pathogénicité à un outil appelé système de sécrétion de type III (SSTT). Fonctionnant comme une seringue, il injecte dans les cellules de l’hôte des toxines nommées Yops (Yersinia Outer Proteins), qui exercent de multiples activités inhibitrices sur les mécanismes cellulaires de la réponse immunitaire innée et peuvent causer la mort des cellules. En 2019, il a été rapporté que le SSTT produirait des N-formylpeptides qui sont alors libérés dans le milieu extracellulaire et qui attirent à la bactérie les cellules du système immunitaire portant FPR1, un récepteur de signaux chimiotactiques (comme CCR5). De plus, FPR1 se lierait à la protéine bactérienne LcrV présente à l’embout de la seringue du SSTT, du fait d’une similarité avec le ligand habituel de FPR1. Cela favoriserait les interactions bactéries-cellules, d’où le surnom de « récepteur de la peste », améliorant ainsi l’injection des Yops et la neutralisation des cellules du système immunitaire. L’étude montrait aussi que des souris déficientes en FPR1 avaient un meilleur taux de survie à l’infection par Y. pestis (36). Chez les humains, la mutation FPR1R190W protège les neutrophiles sanguins contre Y. pestis. Cette mutation a donc pu être sélectionnée lors des épidémies de peste.
FPR1 est également impliqué dans d’autres pathologies. Parmi les plus de 200 mutations ponctuelles (SNP, single nucleotide polymorphism) connues dans le gène, certaines ont été associées à la sensibilité à des maladies comme la dégénérescence maculaire ou le cancer de l’estomac. L’interaction des cellules cancéreuses avec les cellules dendritiques, chefs d’orchestre des réponses immunitaires adaptatives, nécessite un FPR1 fonctionnel. Ce récepteur est ainsi nécessaire à l’immunité humaine anticancéreuse. De ce fait, les auteurs de l’étude ont proposé que les principaux SNP de FPR1 ont pu être sélectionnés par les maladies infectieuses, dont la peste, mais au détriment de la résistance des individus au cancer. Ils notent aussi que, du fait que les cancers se développent généralement tard dans la vie, il est improbable qu’à l’inverse, ce type de maladie exerce une sélection positive sur FPR1 (36).
Outre FPR1, il existe d’autres récepteurs cellulaires eucaryotes pour Y. pestis. Ce sont principalement des lectines (protéines liant des sucres) : SIGNR1/CD2099b (37) ou la Langerine/CD207 (38) se lient ainsi aux oligosaccharides du lipopolysaccharide (LPS) composant la paroi bactérienne. La lectine DEC-205 ne reconnaît pas un sucre de Y. pestis, mais sa protéase Pla, un facteur de virulence essentiel pour la bactérie (39). Toutefois, du fait que ces lectines ne fixent pas seulement les antigènes de Y. pestis, il serait difficile de dire quel microbe aurait exercé la pression de sélection amenant à leur perte, et cela n’a pas été étudié.
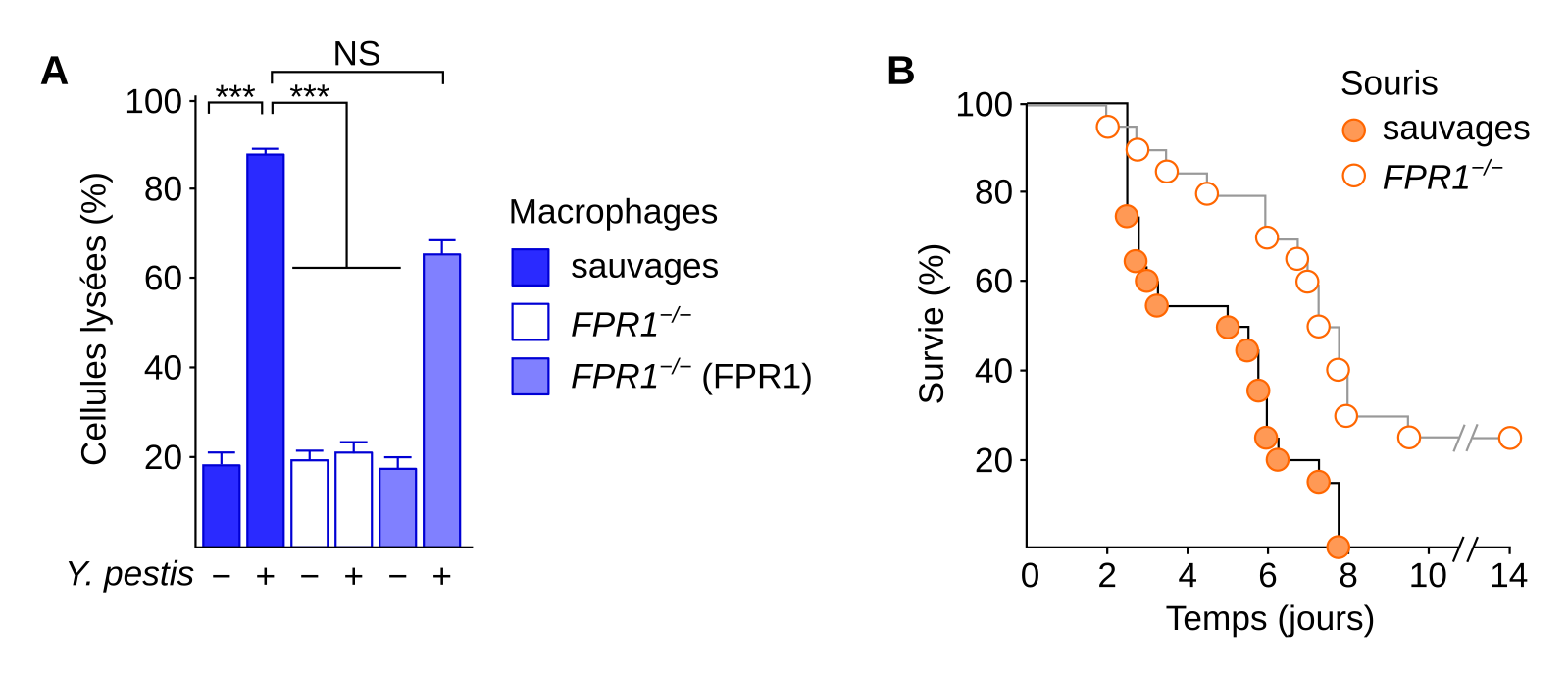
A. Différents types de macrophages (sauvages, mutants pour le gène FPR1 ou mutants mais transfectés avec un plasmide codant FPR1) sont infectés (+) ou non (−) par Yersinia pestis. On mesure le pourcentage de cellules lysées. Les données sont représentées sous la forme de moyennes associées à des barres d’erreur représentant les erreurs types de la moyenne. *** : p < 0,001.
B. Des souris sauvages ou mutantes pour le gène FPR1 sont inoculées avec 600 unités formant colonies de Y. pestis. La survie est suivie sur 14 jours. n = 10 mâles et 10 femelles pour chaque condition.
NOD2 et la maladie de Crohn
L’immunité innée s’appuie sur un jeu limité de récepteurs de reconnaissance de motifs moléculaires (pattern-recognition receptors ; PRR) qui reconnaissent des molécules microbiennes conservées car essentielles à leur physiologie (40). Ces antigènes microbiens sont appelés « motifs moléculaires associés aux pathogènes » (ou PAMP, pour pathogen-associated molecular patterns) et leur reconnaissance par plusieurs classes de PRR active les cellules sentinelles et déclenche plusieurs réponses antipathogènes distinctes (41). Les PRR de la famille NOD (nucleotide-binding oligomerization domain) lient des PAMP cytosoliques. En particulier, NOD2 (NLRC3) reconnaît des muropeptides intracellulaires dérivés du peptidoglycane des bactéries issu des parois des bactéries, et il déclenche des réponses inflammatoires en activant des signaux NF-?B, MAP-kinases, interférons de type 1 ainsi que le processus d’autophagie.
Bien qu’il soit surtout connu pour son rôle dans la détection de nombreux pathogènes, des variations du gène NOD2 prédisposent le plus fortement et spécifiquement à la maladie de Crohn, l’une des deux formes majeures de maladie inflammatoire chronique de l’intestin. Contrairement à ce qui est observé en Asie et en Afrique, jusqu’à 30 à 50 % des patients de Crohn d’origine européenne et 10 % des témoins sains portent au moins une mutation perte de fonction affectant NOD2. Y. pseudotuberculosis, l’espèce la plus proche de Y. pestis, exploite la voie NOD2 par l’intermédiaire de sa toxine YopJ, causant ainsi l’inflammation qui perturbe la fonction de barrière de l’épithélium intestinal. Ainsi, les souris mutées déficientes en NOD2 sont plus résistantes à l’infection orale (42). Parce que Y. pestis possède le même YopJ, la mutation de NOD2 pourrait avoir favorisé la survie à la peste. Par une méta-analyse des résultats publiés, Dumay et ses collègues ont en effet mis en évidence des associations entre la fréquence actuelle de mutations de NOD2 et l’exposition passée des populations à la peste dans les pays d’Europe et du bassin méditerranéen, suggérant que les niveaux élevés de mutations seraient des conséquences négatives des pandémies passées (43).
La relation entre la maladie de Crohn et les Yersinia entéropathogènes (Y. enterocolitica & Y. pseudotuberculosis) n’est toutefois toujours pas claire. L’infection par ces pathogènes modérés a été observée chez les patients atteints de Crohn, et il a été proposé qu’ils y contribueraient en déclenchant une inflammation via NOD2. Cette possibilité s’oppose à l’idée que la mutation de NOD2 favorise la maladie de Crohn, puisqu’elle protège contre l’infection par les Yersinia (42). Une explication possible serait que YopJ n’inhibe pas mais redirige les signaux de NOD2, réduisant l’activation de la voie inflammatoire classique NF-KB pour favoriser une autre : l’activation de la caspase-1, entrainant le processus de mort cellulaire programmée et inflammatoire nommé pyroptose1 (Fig. 1 ; (42)). Par des mécanismes restant à identifier, la mutation de NOD2 pourrait ainsi favoriser une forme plus chronique d’inflammation intestinale par les Yersinia, ouvrant la voie à la maladie de Crohn.
Le gène MEFV de la pyrine et la fièvre méditerranéenne familiale
Plusieurs maladies inflammatoires monogéniques sont dues à des mutations dans des gènes impliqués dans des mécanismes inflammatoires. Le gène muté responsable de la fièvre méditerranéenne familiale, nommé MEFV, encode la pyrine, un récepteur spécifiquement exprimé par les phagocytes (monocytes-macrophages et neutrophiles) (44). La pyrine appartient à la famille des récepteurs NOD-like (NLR) qui détectent des molécules microbiennes dans le cytosol, directement ou grâce à un capteur spécifique. La pyrine a la particularité unique de s’activer par déphosphorylation lorsque les GTPases Rho sont inactivées par des toxines microbiennes. La pyrine s’oligomérise alors en l’une des plateformes nommées inflammasome ; elle recrute alors des caspases inflammatoires comme la caspase 1 (Fig. 1), ce qui déclenche la pyroptose (45). Ces processus font partie des défenses immunitaires innées de l’hôte et sont essentielles à sa protection contre les bactéries et les virus. Chez les sujets atteints de fièvre méditerranéenne familiale, la mutation du gène MEFV codant la pyrine entraîne une inflammation excessive qui cause des épisodes de fièvre, sérite, arthrite et éruptions cutanées ainsi qu’une tendance à l’amyloïdose.
De manière très intéressante, les Yersinia pathogènes constituent les seules bactéries capables d’inhiber la pyrine. Cette inhibition est due à la toxine injectable YopM, qui détourne des protéines kinases qui maintiennent l’état phosphorylé (inhibé) de la pyrine, ce qui empêche la formation de l’inflammasome (46). Ce mécanisme contrebalance l’inhibition des GTPases Rho par YopE et YopT, et fait partie des nombreux outils de virulence utilisés par les Yersinia pour échapper aux défenses antimicrobiennes de l’hôte, contribuant ainsi grandement à la virulence.
Il a été suggéré que la peste aurait sélectionné une mutation gain de fonction de MEFV dans la population humaine (46). Dans une publication de 2020, Park et ses collaborateurs donnent une argumentation solide en faveur de cette hypothèse. D’abord, la fréquence de mutation de MEFV dans les populations méditerranéennes est élevée malgré une espérance de vie réduite par rapport à leurs concitoyens. Ils ont observé que, dans la population turque moderne, trois mutations résultaient d’une sélection récente (2 000 ans), en accord avec la possibilité d’une sélection par les deux grandes pandémies de peste. Pour identifier le mécanisme par lequel la mutation favorise la résistance, ils ont montré que dans les macrophages, la fixation de YopM sur la pyrine mutée est diminuée, avec pour résultat une plus faible production de l’IL-1ß. Finalement, à l’aide de souris ayant reçu le gène FEMV humain, ils montrent que deux des mutations sur la pyrine causant la fièvre méditerranéenne familiale confèrent une meilleure résistance à une infection mortelle par Y. pestis (44).
Il y a toujours la possibilité que d’autres infections aient aussi sélectionné positivement les mutations de la pyrine, mais avec tous ces éléments, Park et coll. apportent une des démonstrations les plus convaincantes que la peste a contribué à modeler le génome humain et que les pandémies historiques ont été un des moteurs de la sélection de certains allèles du gène MEFV dans la population méditerranéenne.
Les études d’association pangénomiques
Dans les études décrites ci-dessus, l’attention était focalisée sur un ou plusieurs gènes candidats pour lesquels une association génétique avec le phénotype étudié était recherchée. Les hypothèses sur lesquelles elles étaient fondées étaient formulées grâce à des connaissances a priori sur les mécanismes par lesquels ces gènes pourraient influencer le développement de la maladie. De ce fait, ce type d’approche est limité dans sa capacité à identifier de nouveaux gènes impliqués. Au contraire, une étude d’association pangénomique (en anglais genome-wide association study, GWAS) vise à identifier, sans a priori, des gènes impliqués dans des caractéristiques individuelles, ou traits phénotypiques (dont les maladies). En pratique, ce type d’étude compare une cohorte, c’est-à-dire un grand groupe de sujets, à un groupe témoin proche (en général de mêmes origines et mode de vie) mais différant par le trait étudié, qui est le plus souvent la présence d’une maladie.
Pour comparer les génomes, la première méthode utilisée, avant le séquençage, a été d’analyser les polymorphismes mononucléotidiques (SNP) du génome à l’aide de puces à ADN (environ 500 000 SNP par puce). Cela permet d’identifier sans a priori les allèles significativement sur ou sous-représentés dans un des groupes. Chaque SNP représentant une région du génome, il n’est souvent pas la cause directe de la maladie, mais l’étude en détail des régions identifiées permet ensuite de pointer vers un nombre plus restreint de gènes ou de voies biologiques réellement impliqués dans la maladie (47). Cependant, il a été objecté que les SNP présents sur les puces ne rendent compte que d’une partie de la diversité génétique, le reste étant constitué de variants rares (ayant des fréquences inférieures à 1 %, voire à 1 ‰) ponctuels ou non, que les puces n’identifient pas. Seul le séquençage complet du génome des sujets de l’étude permet de les détecter, ce qui est depuis peu faisable à prix abordable grâce aux méthodes à haut débit (48). Pour augmenter la puissance de telles études, plusieurs paramètres peuvent être mesurés pour chaque sujet ; ce sont souvent des niveaux d’expression de gènes par analyse d’ARN, ou de production de biomarqueurs (par exemple le taux d’insuline pour le diabète).
Parmi les près de 200 000 études pangénomiques répertoriées dans le catalogue international (www.ebi.ac.uk/gwas ; mai 2020), certaines ont analysé l’empreinte laissée sur le génome humain par des maladies infectieuses telles que la lèpre, le choléra, la malaria, la tuberculose, la variole et le sida (3). La peste a fait l’objet de peu d’études.
Les études pangénomiques apportent une considérable puissance d’analyse et permettent de revoir certaines conclusions. Par exemple, une telle étude en 2005 a permis de réexaminer l’idée d’une sélection positive ayant agi sur le locus CCR5. Elle a évalué l’âge de la mutation à 5 000 ans et n’a pas trouvé de preuves de sélection positive de CCR5, suggérant ainsi que ni la peste, ni le sida, ni la variole ne seraient en cause dans la fréquence élevée de la mutation CCR5-∆32.
Études d’association pangénomiques et études d’interactomique hôte-Y. pestis
Parmi les méthodes les plus récentes développées pour étudier les interactions entre les agents pathogènes et leur hôte, celles regroupées sous le nom d’interactomique étudient les contacts au niveau moléculaire et permettent d’identifier les cibles des facteurs de virulence du pathogène. Les bases de données générées par les études d’association pangénomiques et d’interactomique étant publiques, elles peuvent être combinées afin d’identifier des protéines humaines issues de gènes polymorphiques, et qui interagissent avec Y. pestis. Par une telle approche, C. J. Patel a été amené à conclure que la peste avait exercé une pression de sélection sur plusieurs centaines de gènes (50). Toutefois, cette méthode ne distingue pas encore suffisamment les effets spécifiques de Y. pestis de ceux d’autres bactéries avec lesquelles elle partage des gènes communs, et ne peut pas dire si les variations géniques identifiées ont des conséquences sur la sensibilité à la peste.
Cependant, comme toutes les études génétiques, elle ne permet pas non plus de préciser le mécanisme pathologique à l’origine de la sélection, et devrait être complétée par des tests fonctionnels (délétion de gènes de virulence de Y. pestis, étude de sous-populations humaines présentant des allèles mutés du gène, etc.).
Sélection de récepteurs Toll-like (TLR) par la peste
Netea et ses collègues ont émis l’hypothèse que la forte pression de sélection exercée par les pandémies des siècles passés (parmi lesquelles la peste a joué un rôle majeur) a pu mener à une évolution convergente et à la sélection d’allèles signatures dans des populations variées, indépendamment du contexte génétique de départ (51). Ils ont exploité une particularité de l’histoire de Roumanie : l’immigration des Roms (ou Tziganes) depuis le nord de l’Inde il y a un millénaire pour vivre depuis côte à côte avec les Européens, sans unions entre les deux peuples, maintenant ainsi des patrimoines génétiques distincts. Les auteurs ont examiné les génomes à la recherche d’allèles surreprésentés en Roumanie mais pas chez les Indiens du Nord, dont les ancêtres n’ont pas été exposés à la peste. Bien que les Roms soient encore génétiquement similaires aux Indiens, certains SNP avaient été sélectionnés positivement chez les Roms. Parmi ceux-ci, trois gènes codaient des récepteurs Toll-like (TLR), impliqués dans le fonctionnement du système immunitaire : TLR1, TLR6 et TLR10.
Les récepteurs Toll-like forment une famille de récepteurs de reconnaissance de motifs moléculaires, qui sont ancrés dans les membranes. Chez les humains, les TLR3, 7, 8 et 9 détectent les ADN et ARN et sont spécialisés dans la reconnaissance des virus dans les compartiments internes de la cellule. Les TLR1, 2, 4, 5, 6 et 10 sont généralement exposés à la surface de la cellule et détectent d’autres cibles telles que des glycolipides, des lipopeptides et la flagelline, présents chez une grande variété d’organismes dont les bactéries, les parasites eucaryotes et les champignons.
Afin de tester la possibilité que les allèles des gènes TLR mis en évidence grâce aux SNP aient été sélectionnés sous la pression exercée par Y. pestis, les auteurs ont utilisé la bactérie pour stimuler des cellules de sujets européens exprimant diversement ces allèles de TLR. Chez les Roms possédant les variants des gènes TLR, la production de cytokines était significativement augmentée par rapport aux populations ne les ayant pas, montrant que la variabilité génétique est vraiment déterminante pour les interactions entre Y. pestis et ces récepteurs. Ces observations suggèrent fortement que la peste a favorisé l’évolution des fréquences alléliques de ces gènes par sélection naturelle. Pour consolider cette conclusion, il serait utile de déterminer si cette variation génétique influence la réponse à d’autres pathogènes qui auraient eux aussi pu avoir favorisé ces allèles.

Le génotype de 101 individus européens a été établi vis-à-vis de trois polymorphismes mononucléotidiques (SNP) au niveau des gènes TLR1, TLR6 et TLR10. Pour chaque SNP, les individus sont soit homozygotes sauvages (wt), soit hétérozygotes (he) soit homozygotes mutants (ho). La production de cytokines par les cellules mononucléées sanguines périphériques (lymphocytes et monocytes) de chaque individu est mesurée en réponse à une stimulation par Y. pestis. Les données sont représentées sous la forme de moyennes associées à des barres d’erreur représentant les erreurs types de la moyenne. * p = 0,05 ; ** p = 0,01 ; *** p = 0,001.
Une étude plus ancienne conduite en 2009 par L. Barreiro a montré que la diversité génétique pour la plupart des TLR était plus élevée chez les Africains que chez les Européens et les Asiatiques de l’Est (52). En particulier, la région TLR1-6-10 a été la cible d’une sélection positive récente chez les non-Africains. Parce que la plus grande part de l’Afrique a été épargnée par la peste, il est possible que ces haplotypes aient été sélectionnés par les grandes pandémies comme la peste parce qu’elles conféraient un avantage contre l’infection.
Du fait de leur rôle important dans l’immunité innée, on pouvait s’attendre à ce que la peste ait exercé une pression de sélection sur les TLR. Ces récepteurs sont essentiels à la défense de l’hôte contre de nombreuses bactéries et Y. pestis a développé des mécanismes d’évasion pour échapper aux défenses immunitaires. Depaolo a montré que l’hétérodimère TLR1/6 porté – entre autres – par les cellules dendritiques (DC, sentinelles du système immunitaire) reconnaissait l’antigène LcrV (comme FPR1, voir supra) et que cela stimulait la production de l’IL-10, une cytokine anti-inflammatoire (53). La fonction normale des cellules dendritiques est d’activer les lymphocytes T, mais le signal donné par LcrV/TLR6 les avait poussées à générer des cellules T tolérantes. De plus, les souris déficientes en TLR6 étaient plus résistantes à la peste.
Comme pour NOD2 ou FPR1, un inconvénient de la sélection de TLR variants semble être que les Européens contemporains sont plus sujets à développer des maladies auto-immunes ou inflammatoires que ceux dont les ancêtres n’ont jamais subi la peste. En effet, les variations de TLR1 et TLR10 ont été associées avec la sarcoïdose et une forme de Crohn indépendante de NOD2 (55, 56).
Impact de la peste sur l’évolution phénotypique du génome humain
Alors que l’étude par Laayouni et ses collèguesétait axée sur des populations caucasiennes de Roumanie, aucune équipe n’a encore comparé des populations plus distantes différant par leur exposition historique à la peste. Un projet est en cours, dirigé par le Dr Luis Barreiro de l’université de Chicago, et auquel notre équipe à l’Institut Pasteur participe (57). Il consiste en une comparaison de sujets Américains d’origine africaine ou européenne. En effet, alors que les deux premières pandémies de peste ont ravagé l’Europe et l’Asie, les populations africaines n’ont pas subi de telles mortalités (58), et la comparaison de leur réponse immunitaire contre Y. pestis testée in vitro devrait permettre de déterminer si l’exposition historique de ces populations à la peste est réellement responsable de la sélection de profils de locus de traits quantitatifs d’expression (eQTL1) distincts, reflétant une sélection naturelle.
Dans une première étude rapportée par J.F. Brinkworth, des Américains d’origine africaine ou européenne ont été comparés (59). Lorsque les macrophages étaient stimulés par du Y. pestis tué, des différences d’expression ont été observées pour plusieurs familles de gènes, dont certains impliqués dans l’inflammation, suggérant des différences de régulation de voies de signalisation de la réponse immunitaire entre les deux groupes.
Dans l’étude en cours, une approche similaire mais plus ambitieuse est suivie avec plus de sujets, et les macrophages sont exposés à du Y. pestis tout à fait virulent et vivant afin d’utiliser le niveau d’expression de multiples gènes pour définir des profils distincts entre les deux groupes d’individus. Nous espérons non seulement caractériser la variabilité interindividuelle et entre populations de la réponse immunitaire à l’infection, mais aussi dresser la carte génétique des eQTL associés à cette variation.
De plus, en collaboration de H. Poinar (Université Mc Master, Hamilton, Canada), les loci identifiés chez les sujets contemporains seront examinés dans les ADN anciens provenant de squelettes d’individus ayant vécu avant, pendant et après la grande peste noire médiévale. Cela devrait permettre d’identifier les loci génétiques portant la signature d’une sélection positive par Y. pestis, révélée par une augmentation de la fréquence d’allèles conférant la résistance ou une protection partielle contre Y. pestis chez des gens ayant vécu après la pandémie, comparés à ceux ayant vécu avant.
Covid-19, peste et sélection naturelle dans les populations humaines
La pandémie de Covid-19 actuelle est souvent comparée à la peste, et ce malgré le fait que les deux microbes soient très différents (un virus et une bactérie). Les deux maladies ont en commun des formes respiratoires potentiellement mortelles, une contagiosité importante, et une dissémination internationale accompagnée de réactions de peur et de mesures de confinement. Pour ce qui est de la sélection naturelle qu’elles exercent sur l’humanité, un facteur limitant le pouvoir sélectif de la Covid-19 est l’âge moyen élevé des victimes, qui sont donc peu concernées par le risque de ne pas avoir de descendance. La situation est différente pour la peste qui touche des sujets de tous âges. De plus, le coronavirus exercera une pression de sélection à la mesure de ses conséquences sur la survie et la démographie des populations, et heureusement la Covid-19 est encore loin d’avoir causé les 200 millions de morts estimées pour les deux grandes pandémies de peste.
Les pressions de sélection exercées par les maladies infectieuses ne sont probablement plus si fortes que dans le passé. En effet, la découverte de traitements préventifs et curatifs (en particulier des vaccins et des antibiotiques) a permis, au moins pour les populations y ayant accès, de réduire la mortalité due aux infections. Toutefois cette situation pourrait n’être qu’une parenthèse dans l’histoire humaine du fait de l’émergence constante de bactéries résistantes aux antibiotiques, dont des Y. pestis.
La pression de sélection sur Yersinia pestis et les animaux réservoirs
Apparition de Yersinia pestis : sélection d’un tueur en série
Dans la lutte permanente entre les microbes et leurs hôtes, les pathogènes évoluent constamment et trouvent de nouvelles manières pour échapper au système immunitaire et infecter leurs cibles. Cette évolution est d’une certaine manière en miroir à celle de l’hôte, mais l’échelle de temps pour la sélection naturelle des micro-organismes est considérablement plus rapide que celle des organismes pluricellulaires. Elle est en effet facilitée par leur grande vitesse de reproduction et l’existence de multiples mécanismes de remodelage de leurs éléments génétiques, rendant faciles l’acquisition ou la perte de gènes. Y. pestis est ainsi une espèce apparue il y a moins de 10 000 ans, lorsqu’un clone s’est séparé de l’espèce Yersinia pseudotuberculosis bien moins virulente. En fait, Y. pestis reste génétiquement une Yersinia pseudotuberculosis, mais est néanmoins considérée comme une espèce distincte du fait de sa pathogénicité extrême (60). En effet, Y. pestis n’a acquis que peu de gènes nouveaux (8 régions chromosomiques et deux plasmides) et la transformation a impliqué plus d’inactivation de fonctions par perte de matériel génétique (13 régions) et par inactivation de séquences codantes (208 pseudogènes) (61).
L’ancêtre modérément pathogène de Y. pestis, proche de certaines Y. pseudotuberculosis contemporaines, possédait déjà un certain nombre de facteurs pour la virulence chez les mammifères, dont les plus notables sont l’îlot de haute pathogénicité1 chromosomique codant le système yersiniabactine de capture de fer, et le système de sécrétion de type III, codé sur le plasmide de virulence pYV/pCD1. Cet ancêtre avait aussi déjà le tropisme vers les organes lymphoïdes que Y. pestis a gardé. Le changement de mode de transmission, de l’orofécal à un mode transmis par insecte vecteur (la puce) a nécessité seulement quelques changements génétiques. La Yersinia pestis la plus ancestrale fraîchement mutée est probablement passée d’un mammifère septicémique infecté par voie orale à une puce venue se nourrir, et a ainsi été sélectionnée. Les mutations nécessaires incluent l’acquisition du gène ymt, codant une phospholipase D protégeant la bactérie dans la puce, des pertes de fonctions permettant la formation d’un biofilm dans l’intestin de la puce, et l’inactivation du gène de l’uréase ureD, par laquelle Y. pestis est devenu non toxique pour la puce (62-64). La nouvelle capacité de la bactérie à infecter les puces lui a ouvert un accès direct à la peau lors de la piqûre, causant la peste bubonique et refermant le nouveau cycle dans lequel le mammifère hôte est le plus souvent un rongeur, et parfois l’Homme.
L’évolution ayant mené àY. pestis est aussi passée par l’acquisition de deux plasmides pFra/pMT1 et pPla/pPCP1. Le grand plasmide pFra comporte le gène ymt, de nombreux gènes aux fonctions inconnues, ainsi que l’opéron caf, qui produit l’antigène pseudo-capsulaire F1, qui n’est pas essentiel à la virulence. À l’inverse, le petit plasmide pPla contient peu de gènes et est important pour la virulence. Cela est dû au fait que la protéase Pla, activant le plasminogène, contribue à la progression de la bactérie dans la peau et dans les poumons (65, 66), bien que son introduction artificielle chez Y. pseudotuberculosis n’augmente pas sa virulence (67). Plus récemment, la séquence du gène pla a encore varié et le SNP T259, qui optimise l’activité protéasique de Pla, a été sélectionné chez toutes les souches de Y. pestis modernes. Cette modification n’est pas nécessaire à la peste pulmonaire mais est importante pour la peste bubonique (68).
La perte de nombreuses fonctions devenues non nécessaires est supposée avoir eu lieu après le changement de mode de transmission. Par exemple de nombreux gènes impliqués dans le métabolisme chez l’ancêtre commun de Y. pseudotuberculosis et Y. pestis sont devenus des pseudogènes chez cette dernière, ce qui s’explique probablement par le fait que son cycle ne passe plus par l’environnement et que la bactérie n’a plus besoin des gènes qui permettent d’y survivre (64). Plus important, l’inactivation de certains gènes a augmenté la virulence en permettant l’évasion immunitaire. Par exemple, la perte du flagelle (et donc de la motilité) lui a permis d’échapper au TLR5 de l’hôte qui détecte la flagelline. Aussi, la mutation faisant de LpxL un pseudogène a causé l’hypoacylation du LPS à la température de l’hôte (37 °C), ce qui évite la reconnaissance du LPS par TLR4 (54) (Fig. 2). De telles modifications ont augmenté l’adaptation de Y. pestis à son nouvel environnement et ont été sélectionnées positivement chez les Y. pestis contemporaines.
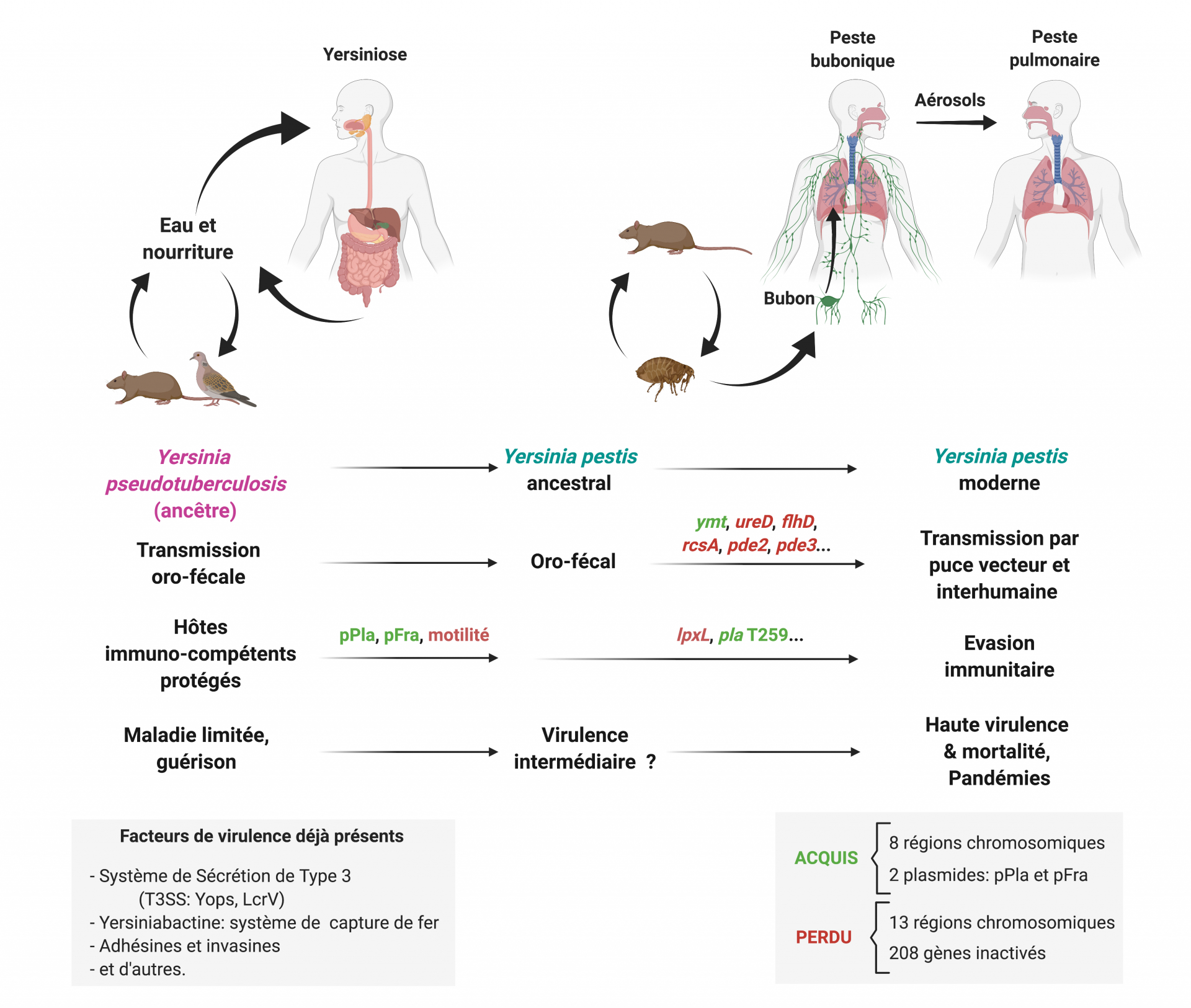
Représentation schématique des étapes successives par lesquelles la sélection de modifications génétiques chez une bactérie faiblement entéropathogène a permis l’émergence d’un pathogène à transmission vectorielle, capable d’infecter les humains par la peau ou les poumons, à échapper à leur système immunitaire, et à provoquer la peste, une maladie hautement mortelle, dont la transmission facile a été à l’origine de pandémies majeures. Les flèches noires indiquent les routes de contamination. Les gènes acquis sont notes en rouge, ceux perdus ou inactivés sont en vert. pFra et pPla sont les plasmides acquis par Y. pestis.
Pression de sélection sur les animaux réservoirs
Alors que les deux autres espèces de Yersinia pathogènes pour les mammifères (Y. pseudotuberculosis et Y. enterocolitica) sont des entéropathogènes ayant un cycle infectieux oro-fécal, Y. pestis a développé un cycle passant par la puce, et ses hôtes mammifères, principalement rongeurs (64). En conséquence, la peste est essentiellement une maladie des animaux (maladie zoonotique) et de ce fait la pression de sélection sur ces animaux est certainement plus forte que sur l’Homme.
Les rongeurs
Commensal de l’Homme, le rat Rattus rattus représente aussi l’une des premières victimes des épidémies de peste, au point que la présence de rat morts en est devenue un signe annonciateur de la calamité (58). La pression de sélection par la peste est donc la plus forte sur ces animaux. Le rôle joué par le rat a fait l’objet de plusieurs études à Madagascar, pays déclarant actuellement le plus de cas de peste par an (69). Dans les zones d’endémie, les rats sont plus résistants, en accord avec un rôle plausible de réservoir, et des mutations conférant cette résistance pourraient avoir été sélectionnées positivement. Une mutation de CCR5 a été recherchée chez les rats résistants comparés à ceux de zones sans peste, mais aucune différence de fréquence n’a été trouvée (70). Vingt-deux autres marqueurs génétiques associés à la résistance ont été identifiés par une approche génomique chez le rat de zone endémique (hauts plateaux), dont deux ont été associés à la résistance lors de tests d’infection expérimentale, probablement sélectionnés par la peste (71).
Comme les rats, les souris sont le plus souvent sensibles à la peste, et toutes les lignées murines de laboratoire habituelles (Mus musculus) sont sensibles. Pourtant, la lignée de souris SEG de l’espèce Mus spretus (la souris d’Afrique du Nord) résiste à la peste bubonique (72). Cette lignée a été capturée initialement en Espagne, un pays très exposé historiquement à la peste. Les SEG pourraient donc être des descendantes de survivantes sélectionnées positivement.
L’analyse de leurs mécanismes immunitaires durant l’infection a montré un recrutement plus rapide des cellules de l’immunité innée (73), ainsi qu’une capacité forte de ces cellules à produire une réponse inflammatoire et à résister à la mort induite par la bactérie (74). Les gènes impliqués n’ont pas pu être précisément identifiés, mais par la méthode des QTL appliquée à des croisements répétés avec des souris sensibles (backcross), quatre loci chromosomiques importants ont été associés à la résistance (72, 75). Qu’un nombre limité de gènes portent la capacité de résistance va dans le sens de l’hypothèse d’une sélection génétique par la maladie au cours des pandémies ayant ravagé le pourtour méditerranéen.
En foyer d’épidémie, d’autres petits mammifères constituant le réservoir de la bactérie pourraient être plus résistants du fait qu’ils auraient historiquement subi la pression de sélection. Des campagnols résistants ont ainsi été décrits (76) et à Madagascar les musaraignes, également résistantes à la peste, sont un des réservoirs identifiés de la bactérie (77). Il serait intéressant de chercher des différences génétiques avec les mêmes espèces prises en zone épargnée par la peste.
Les chiens et chats, hôtes occasionnels
Tous les mammifères ne présentent pas la même sensibilité à la peste. Un exemple frappant est que les chats sont très sensibles alors que les chiens sont nettement plus résistants. Les chats constituent ainsi une source de contamination humaine plus fréquente que les chiens (78). L’une et l’autre espèce carnivore étant exposées à consommer des rongeurs malades de peste, cette différence suggère un avantage d’origine génétique chez les canidés. Dans les zones d’endémie pesteuse, les chiens résistent bien et ont ainsi été utilisés comme sentinelles de la maladie par des tests de sérologie, puisqu’ils développent des anticorps contre la bactérie (79). De manière notable, tous les canidés (mais pas les félins) ont perdu le gène FPR1 (« récepteur de la peste », voir 3.1), et cela pourrait expliquer leur résistance à la maladie (36). Toutefois, la perte de FPR1 chez les canidés est un évènement ancien, qui peut avoir été causé par une bactérie possédant un système de sécrétion de type III, possiblement une Y. pseudotuberculosis, mais qui ne peut pas être attribué à la peste, trop récente.
Remarques de conclusion
Les pandémies de peste du passé, par leur mortalité élevée et leurs conséquences sociales et économiques majeures, ont laissé une empreinte forte sur la démographie et l’histoire de l’humanité. Leurs conséquences sur le génome humain, plus difficiles à cerner et longtemps négligées, sont maintenant en cours d’évaluation. Il y a peu de doutes que la peste ait été un moteur puissant de sélection naturelle pour les populations concernées. Toutefois, elle tient une place tellement exceptionnelle dans la mémoire collective que cela a pu amener à sous-estimer l’influence des autres infections, aiguës ou chroniques ayant aussi pu influencer le génome humain. Ainsi, parmi les nombreuses hypothèses émises concernant la peste, certaines ont été invalidées et d’autres sont encore en cours de clarification.
Avec les méthodes récentes d’analyse des marques de sélection dans le génome, des conclusions plus rigoureuses ont pu être tirées (2, 3). Les études pangénomiques rendent maintenant possible l’identification de gènes inattendus, par exemple ceux dont la participation aux défenses immunitaires n’était pas connue, ou bien dont le rôle était trop indirect pour avoir été décrit jusqu’ici. Une fois de tels gènes identifiés, il est nécessaire de prouver que l’allèle mutant trouvé favorise la survie à l’infection par Yersinia pestis. Comme cela a été fait avec succès pour les gènes codant NOD2, FPR1 ou la pyrine, cette démonstration fonctionnelle s’obtient en reproduisant l’allèle étudié chez l’animal (surtout la souris), ou en identifiant les conséquences d’un allèle mutant naturel chez l’humain. Enfin, les analyses d’ADN ancien, par comparaison de génomes avant et après des épisodes de peste, devraient permettre de mettre en évidence de manière claire la sélection d’allèles de résistance chez les survivants.
En enrichissant nos connaissances des interactions hôte-pathogène et des mécanismes de virulence impliqués, les études de génétique humaine contribuent à la compréhension du système immunitaire lui-même, et par là, elles participent à l’amélioration des méthodes de traitement et permettent l’identification préventive des personnes génétiquement les plus sensibles à l’infection.
Remerciements
L’unité de recherche Yersinia de l’Institut Pasteur est un membre du LabEX IBEID (ANR LBX-62 IBEID). M. Carloni a reçu le soutien du programme international de doctorat “Pasteur – Paris University“(PPU). Les auteurs sont aussi reconnaissants à Xavier Montagutelli et Danielle Séhier pour les discussions et suggestions utiles apportées à ce manuscrit. Les figures ont été créées avec BioRender.
Références
1. Darwin, C. R. 1859. The origin of species. John Murray, London.
2. Quintana-Murci, L., and L. B. Barreiro. 2010. The role played by natural selection on Mendelian traits in humans. Ann N Y Acad Sci 1214 : 1-17.
3. Karlsson, E. K., D. P. Kwiatkowski, and P. C. Sabeti. 2014. Natural selection and infectious disease in human populations. Nat Rev Genet 15 : 379-393.
4. Casanova, J. L., and L. Abel. 2013. The genetic theory of infectious diseases : a brief history and selected illustrations. Annu Rev Genomics Hum Genet 14 : 215-243.
5. Campbell, N. A., and J. B. Reece. 2005. The Evolution of Populations. In Biology. 7th Edition. Pearson – Benjamin Cummings. 454-471.
6. Brinkworth, J. F., and L. B. Barreiro. 2014. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr Opin Immunol 31 : 66-78.
7. Barreiro, L. B., and L. Quintana-Murci. 2010. From evolutionary genetics to human immunology : how selection shapes host defence genes. Nat Rev Genet 11 : 17-30.
8. Kwiatkowski, D. P. 2005. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet 77 : 171-192.
9. Andrades Valtuena, A., A. Mittnik, F. M. Key, W. Haak, R. Allmae, A. Belinskij, M. Daubaras, M. Feldman, R. Jankauskas, I. Jankovic, K. Massy, M. Novak, S. Pfrengle, S. Reinhold, M. Slaus, M. A. Spyrou, A. Szecsenyi-Nagy, M. Torv, S. Hansen, K. I. Bos, P. W. Stockhammer, A. Herbig, and J. Krause. 2017. The Stone Age Plague and Its Persistence in Eurasia. Curr Biol 27 : 3683-3691 e3688.
10. Rascovan, N., K. G. Sjogren, K. Kristiansen, R. Nielsen, E. Willerslev, C. Desnues, and S. Rasmussen. 2019. Emergence and Spread of Basal Lineages of Yersinia pestis during the Neolithic Decline. Cell 176 : 295-305 e210.
11. Rasmussen, S., M. E. Allentoft, K. Nielsen, L. Orlando, M. Sikora, K. G. Sjogren, A. G. Pedersen, M. Schubert, A. Van Dam, C. M. Kapel, H. B. Nielsen, S. Brunak, P. Avetisyan, A. Epimakhov, M. V. Khalyapin, A. Gnuni, A. Kriiska, I. Lasak, M. Metspalu, V. Moiseyev, A. Gromov, D. Pokutta, L. Saag, L. Varul, L. Yepiskoposyan, T. Sicheritz-Ponten, R. A. Foley, M. M. Lahr, R. Nielsen, K. Kristiansen, and E. Willerslev. 2015. Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago. Cell 163 : 571-582.
12. Bos, K. I., A. Herbig, J. Sahl, N. Waglechner, M. Fourment, S. A. Forrest, J. Klunk, V. J. Schuenemann, D. Poinar, M. Kuch, G. B. Golding, O. Dutour, P. Keim, D. M. Wagner, E. C. Holmes, J. Krause, and H. N. Poinar. 2016. Eighteenth century Yersinia pestis genomes reveal the long-term persistence of an historical plague focus. Elife 5 : e12994.
13. Audoin-Rouzeau, F. 2003. Les Chemins de la peste : Le rat, la puce et l’homme. Presses Universitaires de Rennes.
14. Kugeler, K. J., J. E. Staples, A. F. Hinckley, K. L. Gage, and P. S. Mead. 2015. Epidemiology of human plague in the United States, 1900-2012. Emerg Infect Dis 21 : 16-22.
15. Bertherat, E. 2019. Plague around the world in 2019. In Weekly Epidemiologic Reports. WHO, ed. WHO. 289-292.
16. Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis--etiologic agent of plague. Clin Microbiol Rev 10 : 35-66.
17. DeWitte, S. N. 2014. Mortality risk and survival in the aftermath of the medieval Black Death. PLoS One 9 : e96513.
18. Cooling, L. 2015. Blood Groups in Infection and Host Susceptibility. Clin Microbiol Rev 28 : 801-870.
19. Dominguez-Bello, M. G., M. E. Perez, M. C. Bortolini, F. M. Salzano, L. R. Pericchi, O. Zambrano-Guzman, and B. Linz. 2008. Amerindian Helicobacter pylori strains go extinct, as european strains expand their host range. PLoS One 3 : e3307.
20. Garratty, G. 2000. Blood groups and disease : a historical perspective. Transfus Med Rev 14 : 291-301.
21. Zauberman, A., Y. Vagima, A. Tidhar, M. Aftalion, D. Gur, S. Rotem, T. Chitlaru, Y. Levy, and E. Mamroud. 2017. Host Iron Nutritional Immunity Induced by a Live Yersinia pestis Vaccine Strain Is Associated with Immediate Protection against Plague. Front Cell Infect Microbiol 7 : 277.
22. Perry, R. D., and J. D. Fetherston. 2011. Yersiniabactin iron uptake : mechanisms and role in Yersinia pestis pathogenesis. Microbes Infect 13 : 808-817.
23. Frank, K. M., O. Schneewind, and W. J. Shieh. 2011. Investigation of a researcher’s death due to septicemic plague. N Engl J Med 364 : 2563-2564.
24. Quenee, L. E., T. M. Hermanas, N. Ciletti, H. Louvel, N. C. Miller, D. Elli, B. Blaylock, A. Mitchell, J. Schroeder, T. Krausz, J. Kanabrocki, and O. Schneewind. 2012. Hereditary hemochromatosis restores the virulence of plague vaccine strains. J Infect Dis 206 : 1050-1058.
25. Moalem, S., M. E. Percy, T. P. Kruck, and R. R. Gelbart. 2002. Epidemic pathogenic selection : an explanation for hereditary hemochromatosis ? Med Hypotheses 59 : 325-329.
26. Samson, M., F. Libert, B. J. Doranz, J. Rucker, C. Liesnard, C. M. Farber, S. Saragosti, C. Lapoumeroulie, J. Cognaux, C. Forceille, G. Muyldermans, C. Verhofstede, G. Burtonboy, M. Georges, T. Imai, S. Rana, Y. Yi, R. J. Smyth, R. G. Collman, R. W. Doms, G. Vassart, and M. Parmentier. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382 : 722-725.
27. Lucotte, G. 2002. Frequencies of 32 base pair deletion of the (Delta 32) allele of the CCR5 HIV-1 co-receptor gene in Caucasians : a comparative analysis. Infect Genet Evol 1 : 201-205.
28. Stephens, J. C., D. E. Reich, D. B. Goldstein, H. D. Shin, M. W. Smith, M. Carrington, C. Winkler, G. A. Huttley, R. Allikmets, L. Schriml, B. Gerrard, M. Malasky, M. D. Ramos, S. Morlot, M. Tzetis, C. Oddoux, F. S. di Giovine, G. Nasioulas, D. Chandler, M. Aseev, M. Hanson, L. Kalaydjieva, D. Glavac, P. Gasparini, E. Kanavakis, M. Claustres, M. Kambouris, H. Ostrer, G. Duff, V. Baranov, H. Sibul, A. Metspalu, D. Goldman, N. Martin, D. Duffy, J. Schmidtke, X. Estivill, S. J. O’Brien, and M. Dean. 1998. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 62 : 1507-1515.
29. Libert, F., P. Cochaux, G. Beckman, M. Samson, M. Aksenova, A. Cao, A. Czeizel, M. Claustres, C. de la Rua, M. Ferrari, C. Ferrec, G. Glover, B. Grinde, S. Guran, V. Kucinskas, J. Lavinha, B. Mercier, G. Ogur, L. Peltonen, C. Rosatelli, M. Schwartz, V. Spitsyn, L. Timar, L. Beckman, M. Parmentier, and G. Vassart. 1998. The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Hum Mol Genet 7 : 399-406.
30. Mecsas, J., G. Franklin, W. A. Kuziel, R. R. Brubaker, S. Falkow, and D. E. Mosier. 2004. Evolutionary genetics : CCR5 mutation and plague protection. Nature 427 : 606.
31. Galvani, A. P., and M. Slatkin. 2003. Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele. Proc Natl Acad Sci U S A 100 : 15276-15279.
32. Hummel, S., D. Schmidt, B. Kremeyer, B. Herrmann, and M. Oppermann. 2005. Detection of the CCR5-Delta32 HIV resistance gene in Bronze Age skeletons. Genes Immun 6 : 371-374.
33. Saleh, M., J. P. Vaillancourt, R. K. Graham, M. Huyck, S. M. Srinivasula, E. S. Alnemri, M. H. Steinberg, V. Nolan, C. T. Baldwin, R. S. Hotchkiss, T. G. Buchman, B. A. Zehnbauer, M. R. Hayden, L. A. Farrer, S. Roy, and D. W. Nicholson. 2004. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429 : 75-79.
34. Ferwerda, B., M. B. McCall, M. C. de Vries, J. Hopman, B. Maiga, A. Dolo, O. Doumbo, M. Daou, D. de Jong, L. A. Joosten, R. A. Tissingh, F. A. Reubsaet, R. Sauerwein, J. W. van der Meer, A. J. van der Ven, and M. G. Netea. 2009. Caspase-12 and the inflammatory response to Yersinia pestis. PLoS One 4 : e6870.
35. Cotsapas, C., B. F. Voight, E. Rossin, K. Lage, B. M. Neale, C. Wallace, G. R. Abecasis, J. C. Barrett, T. Behrens, J. Cho, P. L. De Jager, J. T. Elder, R. R. Graham, P. Gregersen, L. Klareskog, K. A. Siminovitch, D. A. van Heel, C. Wijmenga, J. Worthington, J. A. Todd, D. A. Hafler, S. S. Rich, M. J. Daly, and F. O. N. o. Consortia. 2011. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet 7 : e1002254.
36. Osei-Owusu, P., T. M. Charlton, H. K. Kim, D. Missiakas, and O. Schneewind. 2019. FPR1 is the plague receptor on host immune cells. Nature 574 : 57-62.
37. Yang, K., Y. He, C. G. Park, Y. S. Kang, P. Zhang, Y. Han, Y. Cui, S. Bulgheresi, A. P. Anisimov, S. V. Dentovskaya, X. Ying, L. Jiang, H. Ding, O. A. Njiri, S. Zhang, G. Zheng, L. Xia, B. Kan, X. Wang, H. Jing, M. Yan, W. Li, Y. Wang, X. Xiamu, G. Chen, D. Ma, S. S. Bartra, G. V. Plano, J. D. Klena, R. Yang, M. Skurnik, and T. Chen. 2019. Yersinia pestis Interacts With SIGNR1 (CD209b) for Promoting Host Dissemination and Infection. Front Immunol 10 : 96.
38. Yang, K., C. G. Park, C. Cheong, S. Bulgheresi, S. Zhang, P. Zhang, Y. He, L. Jiang, H. Huang, H. Ding, Y. Wu, S. Wang, L. Zhang, A. Li, L. Xia, S. S. Bartra, G. V. Plano, M. Skurnik, J. D. Klena, and T. Chen. 2015. Host Langerin (CD207) is a receptor for Yersinia pestis phagocytosis and promotes dissemination. Immunol Cell Biol 93 : 815-824.
39. Zhang, S. S., C. G. Park, P. Zhang, S. S. Bartra, G. V. Plano, J. D. Klena, M. Skurnik, B. J. Hinnebusch, and T. Chen. 2008. Plasminogen activator Pla of Yersinia pestis utilizes murine DEC-205 (CD205) as a receptor to promote dissemination. J Biol Chem 283 : 31511-31521.
40. Janeway, C. A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu Rev Immunol 20 : 197-216.
41. Akira, S., S. Uematsu, and O. Takeuchi. 2006. Pathogen recognition and innate immunity. Cell 124 : 783-801.
42. Meinzer, U., F. Barreau, S. Esmiol-Welterlin, C. Jung, C. Villard, T. Leger, S. Ben-Mkaddem, D. Berrebi, M. Dussaillant, Z. Alnabhani, M. Roy, S. Bonacorsi, H. Wolf-Watz, J. Perroy, V. Ollendorff, and J. P. Hugot. 2012. Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe 11 : 337-351.
43. Dumay, A., O. Gergaud, M. Roy, and J. P. Hugot. 2019. Is Crohn’s Disease the Price to Pay Today for Having Survived the Black Death ? J Crohns Colitis 13 : 1318-1322.
44. Park, Y. H., E. F. Remmers, W. Lee, A. K. Ombrello, L. K. Chung, Z. Shilei, D. L. Stone, M. I. Ivanov, N. A. Loeven, K. S. Barron, P. Hoffmann, M. Nehrebecky, Y. Z. Akkaya-Ulum, E. Sag, B. Balci-Peynircioglu, I. Aksentijevich, A. Gul, C. N. Rotimi, H. Chen, J. B. Bliska, S. Ozen, D. L. Kastner, D. Shriner, and J. J. Chae. 2020. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol.
45. Chan, A. H., and K. Schroder. 2019. Inflammasome signaling and regulation of interleukin-1 family cytokines. J Exp Med 217.
46. Chung, L. K., Y. H. Park, Y. Zheng, I. E. Brodsky, P. Hearing, D. L. Kastner, J. J. Chae, and J. B. Bliska. 2016. The Yersinia Virulence Factor YopM Hijacks Host Kinases to Inhibit Type III Effector-Triggered Activation of the Pyrin Inflammasome. Cell Host Microbe 20 : 296-306.
47. National Human Genome Research Institute. 2020. Genome-Wide Association Studies Fact Sheet. NIH, USA.
48. Jordan, B. 2009. [The decline of genome-wide association studies]. Med Sci (Paris) 25 : 537-539.
49. Sabeti, P. C., E. Walsh, S. F. Schaffner, P. Varilly, B. Fry, H. B. Hutcheson, M. Cullen, T. S. Mikkelsen, J. Roy, N. Patterson, R. Cooper, D. Reich, D. Altshuler, S. O’Brien, and E. S. Lander. 2005. The case for selection at CCR5-Delta32. PLoS Biol 3 : e378.
50. Corona, E., L. Wang, D. Ko, and C. J. Patel. 2018. Systematic detection of positive selection in the human-pathogen interactome and lasting effects on infectious disease susceptibility. PLoS One 13 : e0196676.
51. Laayouni, H., M. Oosting, P. Luisi, M. Ioana, S. Alonso, I. Ricano-Ponce, G. Trynka, A. Zhernakova, T. S. Plantinga, S. C. Cheng, J. W. van der Meer, R. Popp, A. Sood, B. K. Thelma, C. Wijmenga, L. A. Joosten, J. Bertranpetit, and M. G. Netea. 2014. Convergent evolution in European and Rroma populations reveals pressure exerted by plague on Toll-like receptors. Proc Natl Acad Sci U S A 111 : 2668-2673.
52. Barreiro, L. B., M. Ben-Ali, H. Quach, G. Laval, E. Patin, J. K. Pickrell, C. Bouchier, M. Tichit, O. Neyrolles, B. Gicquel, J. R. Kidd, K. K. Kidd, A. Alcais, J. Ragimbeau, S. Pellegrini, L. Abel, J. L. Casanova, and L. Quintana-Murci. 2009. Evolutionary dynamics of human Toll-like receptors and their different contributions to host defense. PLoS Genet 5 : e1000562.
53. Depaolo, R. W., F. Tang, I. Kim, M. Han, N. Levin, N. Ciletti, A. Lin, D. Anderson, O. Schneewind, and B. Jabri. 2008. Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4 : 350-361.
54. Montminy, S. W., N. Khan, S. McGrath, M. J. Walkowicz, F. Sharp, J. E. Conlon, K. Fukase, S. Kusumoto, C. Sweet, K. Miyake, S. Akira, R. J. Cotter, J. D. Goguen, and E. Lien. 2006. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nature Immunology 7 : 1066-1073.
55. Abad, C., M. F. Gonzalez-Escribano, L. M. Diaz-Gallo, J. M. Lucena-Soto, J. L. Marquez, E. Leo, C. Crivell, M. Gomez-Garcia, J. Martin, A. Nunez-Roldan, and J. R. Garcia-Lozano. 2011. Association of Toll-like receptor 10 and susceptibility to Crohn’s disease independent of NOD2. Genes Immun 12 : 635-642.
56. Veltkamp, M., C. H. van Moorsel, G. T. Rijkers, H. J. Ruven, and J. C. Grutters. 2012. Genetic variation in the Toll-like receptor gene cluster (TLR10-TLR1-TLR6) influences disease course in sarcoidosis. Tissue Antigens 79 : 25-32.
57. Barreiro, L. B. 2019. Characterizing the impact of Yersinia Pestis to the phenotypic evolution of the human immune system. Grantome.com.
58. Pollitzer, R. 1954. Plague. W.H.O. Monograph Series 22. World Health Organization, Geneva, Switzerland.
59. Brinkworth, J. F., A. Dumaine, V. Yotova, J. C. Grenier, and L. B. Barreiro. 2016. Impact of Yersinia pestis on immune cells variation in humans. In Annual Meeting of the American Association of Physical Anthropologists. M. A. Katzenberg, ed. Wiley Periodicals, Atlanta. 102-103.
60. Achtman, M., K. Zurth, G. Morelli, G. Torrea, A. Guiyoule, and E. Carniel. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci U S A 96 : 14043-14048.
61. Chain, P. S., E. Carniel, F. W. Larimer, J. Lamerdin, P. O. Stoutland, W. M. Regala, A. M. Georgescu, L. M. Vergez, M. L. Land, V. L. Motin, R. R. Brubaker, J. Fowler, J. Hinnebusch, M. Marceau, C. Medigue, M. Simonet, V. Chenal-Francisque, B. Souza, D. Dacheux, J. M. Elliott, A. Derbise, L. J. Hauser, and E. Garcia. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Procedings of the National Academy of Sciences of the United States of America 101 : 13826-13831.
62. Chouikha, I., and B. J. Hinnebusch. 2014. Silencing urease : a key evolutionary step that facilitated the adaptation of Yersinia pestis to the flea-borne transmission route. Proc Natl Acad Sci U S A 111 : 18709-18714.
63. Sun, Y. C., C. O. Jarrett, C. F. Bosio, and B. J. Hinnebusch. 2014. Retracing the evolutionary path that led to flea-borne transmission of Yersinia pestis. Cell Host Microbe 15 : 578-586.
64. Hinnebusch, B. J., C. O. Jarrett, and D. M. Bland. 2017. “Fleaing” the Plague : Adaptations of Yersinia pestis to Its Insect Vector That Lead to Transmission. Annu Rev Microbiol 71 : 215-232.
65. Sodeinde, O. A., Y. V. B. K. Subrahmanyam, K. Stark, T. Quan, Y. D. Bao, and J. D. Goguen. 1992. A surface protease and the invasive character of plague. Science 258 : 1004-1007.
66. Lathem, W. W., P. A. Price, V. L. Miller, and W. E. Goldman. 2007. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science 315 : 509-513.
67. Pouillot, F., A. Derbise, M. Kukkonen, J. Foulon, T. K. Korhonen, and E. Carniel. 2005. Evaluation of O-antigen inactivation on Pla activity and virulence of Yersinia pseudotuberculosis harbouring the pPla plasmid. Microbiology 151 : 3759-3768.
68. Zimbler, D. L., J. A. Schroeder, J. L. Eddy, and W. W. Lathem. 2015. Early emergence of Yersinia pestis as a severe respiratory pathogen. Nat Commun 6 : 7487.
69. WHO. 2019. Plague around the world in 2019. Wkly. Epidemiol. Rec. 94 : 289-292.
70. Tollenaere, C., L. Rahalison, M. Ranjalahy, S. Rahelinirina, J. M. Duplantier, and C. Brouat. 2008. CCR5 polymorphism and plague resistance in natural populations of the black rat in Madagascar. Infect Genet Evol 8 : 891-897.
71. Tollenaere, C., S. Jacquet, S. Ivanova, A. Loiseau, J. M. Duplantier, R. Streiff, and C. Brouat. 2013. Beyond an AFLP genome scan towards the identification of immune genes involved in plague resistance in Rattus rattus from Madagascar. Mol Ecol 22 : 354-367.
72. Blanchet, C., J. Jaubert, E. Carniel, C. Fayolle, G. Milon, M. Szatanik, J. J. Panthier, and X. Montagutelli. 2011. Mus spretus SEG/Pas mice resist virulent Yersinia pestis, under multigenic control. Genes Immun 12 : 23-30.
73. Demeure, C. E., C. Blanchet, C. Fitting, C. Fayolle, H. Khun, M. Szatanik, G. Milon, J.-J. Panthier, J. Jaubert, X. Montagutelli, M. Huerre, J.-M. Cavaillon, and E. Carniel. 2012. Early Systemic Bacterial Dissemination and a Rapid Innate Immune Response Characterize Genetic Resistance to Plague of SEG Mice. The Journal of infectious diseases 205 : 134-143.
74. Pachulec, E., R. B. Abdelwahed Bagga, L. Chevallier, H. O’Donnell, C. Guillas, J. Jaubert, X. Montagutelli, E. Carniel, and C. E. Demeure. 2017. Enhanced Macrophage M1 Polarization and Resistance to Apoptosis Enable Resistance to Plague. J Infect Dis 216 : 761-770.
75. Chevallier, L., C. Blanchet, J. Jaubert, E. Pachulec, C. E. Demeure, E. Carniel, J. J. Panthier, and X. Montagutelli. 2012. Resistance of Mus spretus SEG/Pas mice to virulent Yersinia pestis requires at least four genetic factors. Genes and Immunity 14 : 35-41.
76. Hubbert, W. T., and M. I. Goldenberg. 1970. Natural resistance to plague : genetic basis in the vole (Microtus californicus). Am J Trop Med Hyg 19 : 1015-1019.
77. Rahelinirina, S., M. Rajerison, S. Telfer, C. Savin, E. Carniel, and J. M. Duplantier. 2017. The Asian house shrew Suncus murinus as a reservoir and source of human outbreaks of plague in Madagascar. PLoS Negl Trop Dis 11 : e0006072.
78. Gage, K. L., D. T. Dennis, K. A. Orloski, P. Ettestad, T. L. Brown, P. J. Reynolds, W. J. Pape, C. L. Fritz, L. G. Carter, and J. D. Stein. 2000. Cases of cat-associated human plague in the Western US, 1977-1998. Clin Infect Dis 30 : 893-900.
79. Kilonzo, B. S., N. D. Gisakanyi, and C. A. Sabuni. 1993. Involvement of dogs in plague epidemiology in Tanzania. Serological observations in domestic animals in Lushoto District. Scand J Infect Dis 25 : 503-506.



