L'immunité innée repose notamment sur la reconnaissance de signatures moléculaires portées par des agents pathogènes (PAMP) ou émis par des cellules anormales (DAMP). Ces signaux sont perçus par des récepteurs de reconnaissance de motifs moléculaires (PRR), membranaires ou cytoplasmiques. Si ces récepteurs sont variés, ils déclenchent cependant des voies de signalisation communes qui convergent vers la production de cytokines pro-inflammatoires et d'interférons de type I.
Le corps humain possède son propre répertoire antigénique, les antigènes du soi, qui correspond à l’ensemble des molécules résultant de l’expression de son génome. Par ailleurs, notre organisme lutte contre les infections par des microorganismes (virus, bactéries et parasites eucaryotes, champignons notamment) qui constituent le non soi mais aussi contre le développement de cellules anormales – notamment tumorales – qui constituent le soi modifié. Cette lutte s’opère grâce à un système de défense constitué de deux composantes : l’immunité innée et l’immunité adaptative. Si la réponse adaptative fait intervenir des cellules spécialisées du système immunitaire, l’immunité innée est une réponse précoce développée par la plupart des cellules de l’hôte infecté. Pour les virus, cette réponse cellulaire innée se traduit habituellement par la production et la sécrétion de nombreuses cytokines inflammatoires et immunomodulatrices, dont les interférons de type I (IFN-I) sont les fers de lance. Dans cet article, nous allons principalement nous intéresser aux récepteurs de reconnaissance de motifs moléculaires (PRR) impliqués dans la réponse à une infection virale.
Concepts de PAMP, DAMP et PRR
Jusqu’au début des années 90, la réponse immunitaire était perçue comme une réponse au non-soi, essentiellement associée à la réponse immunitaire adaptative, mais cette conception ne rendait pas compte des mécanismes responsables de la réponse immunitaire innée qui est la première ligne de défense immédiate de l’organisme lors d’une infection ou de dommages cellulaires. C’est Charles Janeway qui, le premier, a formalisé ce problème en imaginant l’existence de récepteurs de reconnaissance de motifs moléculaires (Pattern Recognition Receptors, PRR), capables de reconnaître un ensemble limité de molécules qui signent la présence d’un pathogène, les motifs moléculaires associés aux pathogènes (Pathogen-Associated Molecular Patterns, PAMP). La découverte et la caractérisation d’un type de PRR, les récepteurs Toll-like (TLR), a valu le prix Nobel à Jules Hoffmann et Bruce Beutler en 2011, et a permis de comprendre les mécanismes moléculaires mis en jeu dans l’immunité innée. Plusieurs familles de PRR ont été identifiées et caractérisées depuis, telles que des membres de la famille des RLR (Retinoic acid-inducible gene (RIG)-I-like receptor) et les différents récepteurs à l’ADN cytosoliques (cGAS/STING, IFI16, etc.). Dans les années 90, Polly Matzinger a enrichi ce débat en développant l’idée que toute réponse immunitaire est une réponse à un signal de « danger ». Celui-ci peut être d’origine exogène (PAMP) mais également d’origine endogène quand des molécules normalement séquestrées au niveau intracellulaire sont libérées par les cellules endommagées ou stressées et deviennent alors détectables par le système immunitaire. Ces signaux moléculaires d’origine endogène sont appelés motifs moléculaires associés aux dégâts (Damage-Associated Molecular Patterns ou DAMP) [1-3].
Les PAMP et DAMP principalement impliqués dans l’activation de la sécrétion des interférons de type I sont des acides nucléiques qui se distinguent de l’ADN génomique et des ARN cellulaires par une localisation anormale ou des motifs moléculaires particuliers. Par exemple, les virus produisent des ARN bicaténaires au cours de leur cycle réplicatif, lesquels présentent par ailleurs une extrémité 5’ bi ou triphosphate. En revanche, la plupart des ARN cellulaires sont exprimés sous forme monocaténaire ce qui évite leur reconnaissance par des PRR spécialisés. Par ailleurs, certains longs ARN bicaténaires produits dans les cellules sont associés à des protéines permettant d’éviter leur reconnaissance [4]. Les acides nucléiques endogènes peuvent néanmoins induire la production d’interférons lorsque la cellule subit un stress. Ainsi, les dommages à l’ADN génomique consécutifs à une irradiation, un stress oxydatif ou des erreurs commises par la machinerie de réplication peuvent conduire à l’induction d’interférons de type I. Il a été montré que des fragments d’ADN simple ou double brin libérés à la suite de l’activation de la machinerie de réparation de l’ADN peuvent être reconnus comme des DAMP, notamment lorsque ces fragments fuitent du noyau vers le cytoplasme [5].
Les TLR
Les TLR constituent une des familles de PRR les plus étudiées. Ce sont des récepteurs transmembranaires ayant d’un côté de la membrane un domaine de liaison aux ligands et de l’autre un domaine TIR (Toll/IL-1 resistance) impliqué dans la transmission du signal [6]. Chez l’Homme, 10 TLR ont été identifiés mais seuls certains sont principalement impliqués dans la réponse antivirale.
|
TLR |
Cellules immunitaires |
Localisation cellulaire |
Ligands exogènes (PAMP) |
Ligands endogènes (DAMP) |
Cytokines induites |
|---|---|---|---|---|---|
|
TLR2 (en association avec TLR1 et TLR6) |
CDc, monocytes, neutrophiles, NK, LB, LT |
Membrane plasmique |
Bactéries : peptidoglycane, lipoprotéines, acide lipotéichoïque… Champignons : zymosane |
HSP60, HSP70, Gp96 biglycane, acide hyaluronique, hyaluronane, HMGB1, versicane, urate de sodium |
Cytokines inflammatoires (TNF-α, IL-6, etc.) |
|
TLR3 |
CDc, NK |
Endosomes |
Virus : ARNdb |
ARNm |
Cytokines inflammatoires (TNF-α, IL-6, etc.) et interférons de type I |
|
TLR4 |
CDc, éosinophiles, monocytes, neutrophiles |
Membrane plasmique |
Bactéries : LPS Virus : protéine de fusion du RSV Champignons : mannane Protozoaires : glycoinositolphospholipides |
HSP22, HSP60, HSP70, HSP72, Gp96, acide hyaluronique, hyaluronane, HMGB1, versicane, fibrinogène, fibronectine… |
|
|
TLR7 |
Éosinophiles, monocytes, neutrophiles, CDp, LB |
Endosomes |
Virus : ARNsb |
ARNsb (complexe immun) |
|
|
TLR8 |
CDc, monocytes, neutrophiles, LT, Trég |
Endosomes |
Virus : ARNsb |
ARNsb (complexe immun) |
|
|
TLR9 |
Éosinophiles, monocytes, neutrophiles, CDp, LB |
Endosomes |
Bactéries, virus et protozoaires : ADN CpG |
Complexe immonoglobuline-chromatine, HMGB |
Les TLR7 et 8 sont généralement présents dans les endosomes où ils reconnaissent l’ARN simple brin (ARNsb) extracellulaire lorsque celui-ci est internalisé par la voie endosomale. À ce titre, ils jouent un rôle clé dans l’induction des interférons par les virus à génome ARN comme ceux de l’immunodéficience humaine (VIH) [7], de la stomatite vésiculaire (VSV) [8] et de la grippe. Le TLR7 est surexprimé dans les cellules dendritiques plasmacytoïdes (pDC) et joue un rôle essentiel dans la production des interférons de type I par ces cellules face aux virus. TLR8 est quant à lui exprimé dans les monocytes/macrophages et dans les cellules dendritiques myéloïdes (CDm) [9]. Son activation entraîne à la fois la production des cytokines pro-inflammatoires par la voie NF-ĸB et la production d’interférons de type I.
Comme TLR7, le récepteur TLR9 est fortement exprimé dans les cellules dendritiques plasmacytoïdes et son activation induit la production massive d’interféron α. Ce récepteur reconnaît l’ADN possédant des motifs CpG non-méthylés [10, 11], ce qui limite l’activation par l’ADN génomique des cellules. De plus, TLR9 est localisé dans la voie des endosomes ce qui permet aux cellules dendritiques plasmacytoïdes de différencier l’ADN cellulaire (du soi) éventuellement présent dans le milieu extracellulaire (et qui en principe ne doit pas induire de réponse immunitaire forte), de celui qui est enveloppé au sein d’une particule virale, ce qui favorise son internalisation et la rencontre avec TLR9 dans les voies endosomales.
Enfin, le récepteur TLR3 est impliqué dans la reconnaissance des ARN bicaténaires extracellulaires. TLR3 est exprimé à la fois à la surface et dans les voies endosomales de plusieurs types cellulaires et en particulier les cellules dendritiques conventionnelles, les macrophages mais aussi certains fibroblastes et cellules épithéliales [9].
Contrairement aux TLR7/8/9, les TLR2 et 4 sont essentiellement exprimés à la surface cellulaire et leurs principaux ligands sont des PAMP bactériens comme le lipopolysaccharide (LPS) ou les lipopeptides di ou triacétylés. Cependant, TLR2 et TLR4 sont également capables de reconnaître des protéines virales présentes dans le milieu extracellulaire, notamment des glycoprotéines d’enveloppe [12]. Ainsi, il a été montré que TLR4 reconnaît la protéine de fusion F du virus respiratoire syncytial (VRS) mais également la protéine GP du virus Ebola [13] et la glycoprotéine d’enveloppe du VSV [14]. De même, TLR2 a été impliqué dans la reconnaissance de nombreuses protéines virales comme les glycoprotéines H et B du cytomégalovirus ou les glycoprotéines gH/gL et gB du virus de l’herpès simplex [12].
Une fois activés, les TLR recrutent des protéines adaptatrices, notamment MyD88 (Myeloid Differentiation primary-response gene 88) et TRIF (TIR domain-containing adaptor inducing IFN) [6]. Les différents TLR utilisent des combinaisons différentes d’adaptateurs pour activer leurs gènes cibles, ce qui explique l’induction de profils de gènes différents [11, 17, 18]. La mobilisation de ces adaptateurs conduit à l’activation des kinases IKKε et TBK1 d’une part, responsables de la phosphorylation des facteurs de transcription IRF3/7 et du complexe IKK d’autre part (IKKα/β/γ) conduisant à l’activation du facteur de transcription NF-ĸB (Nuclear Factor Kappa-light-chain-enhancer of activated B cells) [19]. Les protéines IRF3 et IRF7 forment des homo ou hétérodimères qui migrent dans le noyau et induisent l’expression des IFN-α/β et différents gènes stimulés par les interférons (ISG, interferon stimulated genes) en se fixant sur les éléments de réponse IRE (IRF Response Element) [20, 21]. Alors que l’expression des IFN-α repose principalement sur la formation de dimères d’IRF7, l’induction d’IFN-β nécessite le recrutement des dimères d’IRF3/7, NF-ĸB et le complexe ATF2/c-Jun au niveau du promoteur de ce gène [12]. En outre, NF-ĸB joue un rôle très important dans l’activation des cellules effectrices de la réponse immunitaire innée et adaptatives [22], ainsi que dans la sécrétion des cytokines pro-inflammatoires [20].
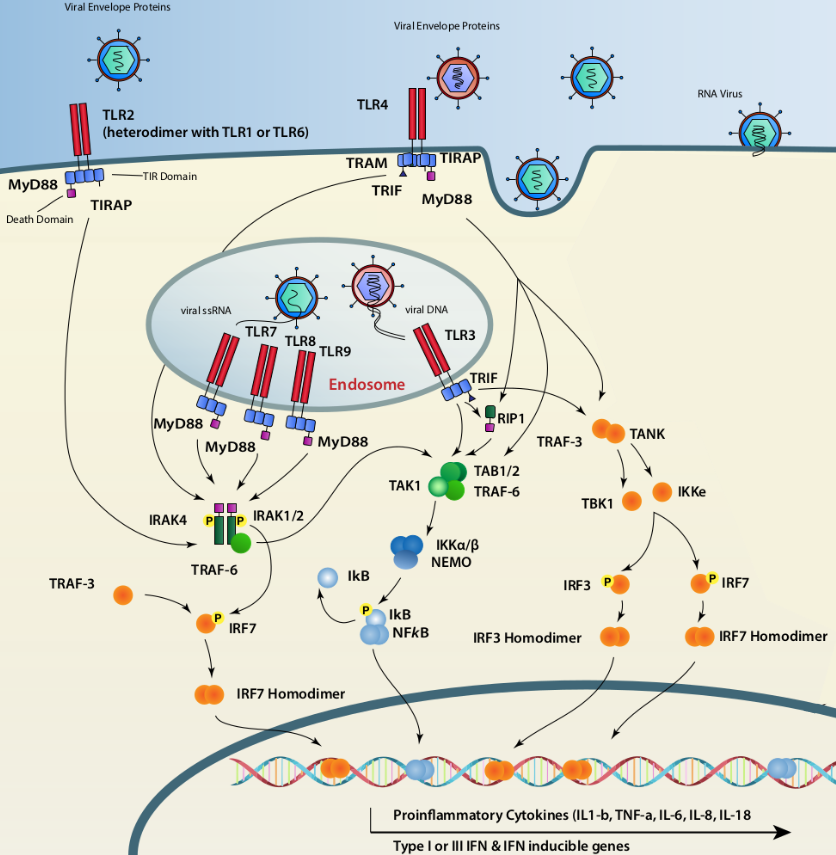
Dans le compartiment endolysosomal, plusieurs TLR peuvent détecter des acides nucléiques différents. TLR3 se lie à un ARNdb, TLR7 détecte un ARNsb et un ARNdb court, TLR8 détecte un ARNsb et TLR9 détecte l’ADN avec une certaine préférence pour les motifs CpG non méthylés. Tous les TLR qui reconnaissent les acides nucléiques, à l’exception de TLR3, transmettent un signal via MyD88 qui, lors de l’activation, forme un complexe de signalisation oligomérique constitué de MyD88, IRAK4 et IRAK1 ou IRAK2, également appelé myddosome. Ce complexe entraîne le recrutement de TRAF6 qui crée une plate-forme d’assemblage, laquelle recrute et active les complexes TAK1 et IKK, ce qui entraîne l’activation de MAPK et de NF-ĸB, respectivement. TLR3, utilise la protéine adaptatrice TRIF qui recrute RIP1 et TRAF6 pour activer les complexes TAK1 et IKK, respectivement. Parallèlement, IKKε et TBK1, sont recrutées via TRAF3, ce qui conduit à la phosphorylation d’IRF3. Adapté d’après Majzoub et coll., 2019.
En résumé, les TLR sont des PRR capables de reconnaître une grande diversité de motifs moléculaires, qu’ils soient associés à des pathogènes (PAMP) ou à des dégâts (DAMP). Cette large gamme de détection est due à la localisation de ces molécules (au niveau de la membrane plasmique, permettant une reconnaissance de motifs extracellulaires, et au niveau des endosomes, permettant une reconnaissance de motifs intracellulaires) et à la variabilité de leur domaine de liaison aux signaux de danger. Le domaine de signalisation est quant à lui relativement conservé d’un TLR à un autre, ce qui explique que les voies activées soient en grande partie communes et aboutissent toutes à la formation de cytokines pro-inflammatoires et, dans la plupart des cas, à la production d’interférons de type I.
TLR et immunité innée chez les Métazoaires
L’immunité innée implique des cellules et molécules communes à un grand nombre d’animaux. Elle se fonde sur la reconnaissance de motifs moléculaires associés aux pathogènes par des récepteurs spécifiques. Les TLR, par exemple, sont des récepteurs qui permettent la reconnaissance de motifs microbiens comme les molécules de la paroi cellulaire bactérienne et l’ADN. À part chez les Spongiaires et chez les Placozoaires, les TLR ont été mis en évidence chez tous les autres Métazoaires [36].
Les récepteurs RLR
Contrairement à la famille des TLR, les membres de la famille des « RIG-I-Like Receptors » (RLR) sont cytosoliques [24]. Cette famille se compose de trois membres bien caractérisés : RIG-I (Retinoic acid-Inducible Gene I, ou DDX58), MDA-5 (Melanoma Differentiation-Associated protein 5, également nommé IFIH1), et LGP2 (Laboratory of Genetics and Physiology 2) [25] (Figure 3). RIG-I détecte les ARN 5’ tri et biphosphates bicaténaires alors que MDA5 reconnaît les ARN bicaténaires de grande taille. Cette spécificité explique que MDA5 et RIG-I soient plus ou moins impliqués dans les réponses à des virus différents [26]. En l’absence d’ARNdb, RIG-I présente une conformation inactive fermée. Dans le cas de MDA5, c’est la multimérisation (ou oligomérisation) le long d’ARN bicaténaires de grande taille qui permet leur activation. Une fois activée par la reconnaissance de leurs ligands ARN respectifs, RIG-I et MDA5 se lient alors à un facteur présent sur la membrane externe des mitochondries : la protéine MAVS (Mitochondrial Antiviral Signaling Protein, également nommée CARDIF, VISA ou IPS1) [27]–[29].
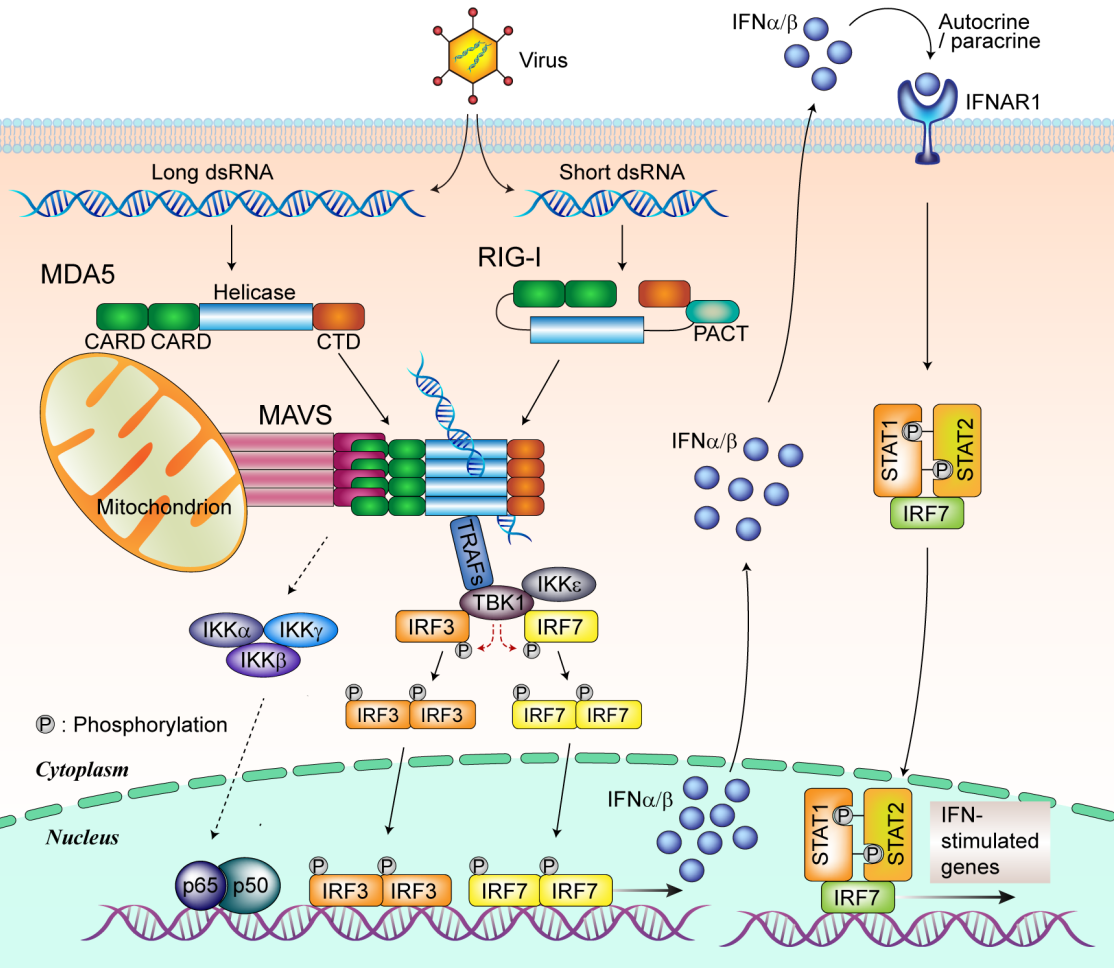
Les récepteurs de type RLR reconnaissent l’ARN génomique ou les intermédiaires de réplication d’ARN de nombreux virus. MDA5 reconnaît les ARN viraux cytoplasmiques double brins (ARNdb), tandis que RIG-I reconnaît les ARNdb viraux courts avec une extrémité 5’ di ou triphosphate. Lors de la reconnaissance des ARN viraux, MDA5 et RIG-I interagissent avec la protéine de signalisation antivirale mitochondriale (MAVS) par interaction de leurs domaines CARD. MAVS module l’activité du facteur de transcription NF-κB via l’activation du complexe IKK (IKK α/β/ɣ). MAVS interagit également avec les protéines TRAF au niveau des mitochondries lors d’une infection virale et induit ensuite le recrutement de TBK1 et IKKε afin de promouvoir la phosphorylation d’IRF3 et IRF7. Une fois phosphorylés, les facteurs IRF3 et IRF7 dimérisent et migrent dans le noyau. Ils se lient alors à des sites de liaison spécifiques présents respectivement dans les promoteurs des gènes IFN-β et IFN-α afin de stimuler la synthèse d’interférons de type I. Une fois sécrétés, ces derniers se lient au récepteur d’IFN-I (IFNAR) et induisent par la suite la phosphorylation de STAT1 et STAT2, conduisant à l’induction de la translocation nucléaire du complexe IRF9 / STAT1 / STAT2 et la transcription des gènes stimulés par les interférons (ISG) [30].
Malgré une redondance fonctionnelle entre RLR et TLR, la reconnaissance par ces récepteurs dépend grandement du pathogène et du type cellulaire. Ainsi, des études sur des souris déficientes en RIG-I et MDA-5 ont révélé que les cellules dendritiques conventionnelles (CDc), les macrophages et les fibroblastes isolés de ces souris présentent une production d’interférons altérée après infection par des virus à ARN, tandis que la production d’interférons est maintenue dans les cellules dendritiques plasmacytoïdes (CDp) [31]. Chez ces dernières, le système TLR apparaît indispensable pour l’induction de la synthèse d’interférons de type I. En revanche, les TLR semblent peu impliqués dans le déclenchement d’une réponse antivirale dans les cellules dendritiques conventionnelles, les macrophages et les fibroblastes. Pour ces types cellulaires, les RLR prennent le relai pour détecter les virus.
Les récepteurs à ADN cytosolique
Un ensemble hétérogène de récepteurs cytosoliques propres à l’ADN double-brin (ADNdb) a été caractérisé récemment pour sa capacité à induire la production des interférons de type I [32] (Figure 4). Ces PRR sont impliqués à la fois dans la détection d’ADN exogène issu d’agents infectieux, virus et bactéries intracellulaires, mais aussi d’ADN endogène d’origine génomique ou mitochondriale [5, 33]. De ce fait, ces récepteurs jouent un rôle clé dans la réponse aux dommages à l’ADN ou au stress métabolique associé à une rupture de la membrane mitochondriale. Parmi ces récepteurs, la protéine cGAS (cyclic GMP-AMP synthase) est l’un des mieux caractérisés [34]. Il s’agit d’une nucléotidyltransférase capable de synthétiser à partir de la guanosine et l’adénosine triphosphate un médiateur intracellulaire, le 2’3’-cGAMP, en réponse à son activation par de l’ADN cytoplasmique. À son tour, le 2’3’-cGAMP se lie au récepteur transmembranaire ancré au réticulum endoplasmique STING (Stimulator of IFN Genes) qui subit un changement de conformation et une translocation vers l’appareil de Golgi et aux endosomes périnucléaires ce qui déclenche une cascade de signalisation conduisant à l’activation d’IRF3 et de NF-ĸB. Comme pour les TLR et RLR, l’activation de ces facteurs de transcription aboutit à la production des interférons de type I et permet d’établir un état antiviral [34].
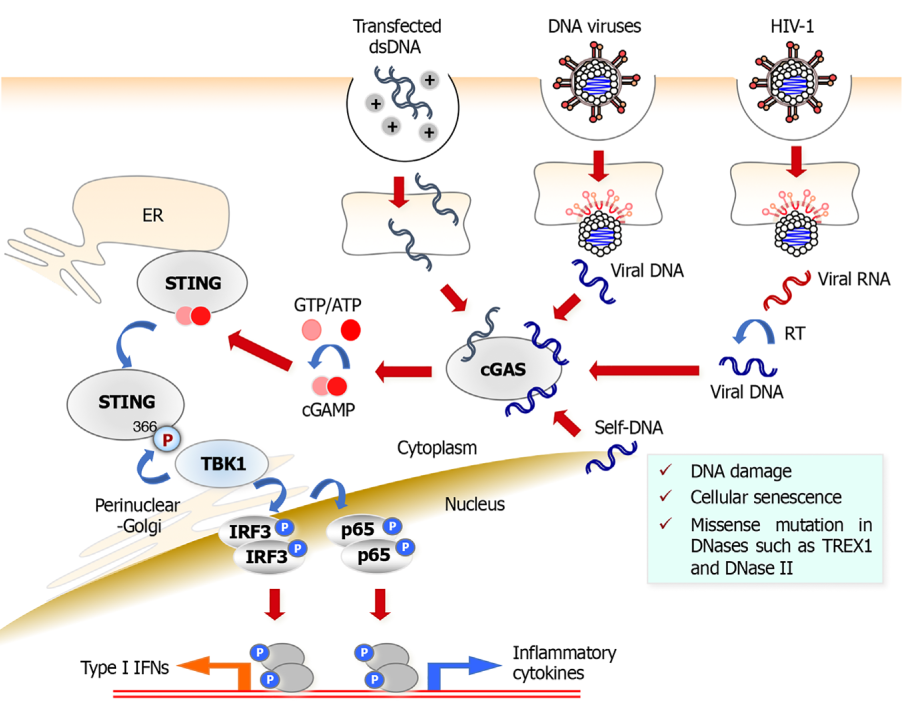
Lors d’une infection par un virus à ADN, le récepteur à l’ADN cytosolique cGAS reconnaît directement l’ADN viral et catalyse la synthèse de cGAMP à partir de GTP et d’ATP intracellulaire. Par la suite, cGAMP se lie à l’adaptateur STING qui est alors activé. cGAS peut également reconnaître l’ADNdb transfecté ou l’ADN viral qui est produit par le VIH-1 lors de l’étape de transcription inverse de l’ARN viral. Une fois lié au cGAMP, STING est transféré du réticulum endoplasmique à l’appareil de Golgi périnucléaire pour former un complexe de signalisation avec la kinase TBK1 et IRF3 pour induire la production d’interférons de type I. STING peut également activer la production de cytokines pro-inflammatoires par l’activation de NF-ĸB (p65). Enfin, cGAS participe aussi à la reconnaissance d’ADN cellulaire endogène tels que les nucléosomes et l’ADN micronucléaire libéré dans le cytoplasme lors de dommages à l’ADN ou au moment de la sénescence ce qui active ainsi la voie STING [35].
D’autres récepteurs ont été identifiés mais sont moins bien étudiés. La plupart utilisent également la voie STING, et parmi ceux-ci nous pouvons citer LRRFIP1, DHX9, DHX36, DDX41, IFI16, DNA-PK et MRE11. Ces trois derniers facteurs sont les mieux caractérisés, mais leur mode d’action et les voies de signalisation qu’ils activent demandent encore à être précisés.
Conclusion
En conclusion la reconnaissance par les cellules de signaux de danger (PAMP et DAMP) met en jeu différents récepteurs de reconnaissance de motifs moléculaires (PRR) : TLR, RLR, et divers récepteurs à l’ADN cytosolique. Ces PRR diffèrent au niveau de leur localisation (membrane plasmique, endosome, cytosol) et au niveau des motifs reconnus (ADN et ARN double brin ou simple brin, protéines, lipides…). Il faut noter que, bien que différentes molécules soient reconnues par ces PRR (protéines, lipides…), la reconnaissance du danger fait le plus souvent intervenir un type de molécule commun à tous les êtres vivants : les acides nucléiques. C’est la différence de structure (simple ou double brin, méthylé ou non, coiffé ou non) et la localisation de ces acides nucléiques qui permet aux cellules de détecter les molécules associées à un danger (matériel génétique issu d’un pathogène, ADN endogène endommagé).
Une fois que les PRR ont reconnu un signal de danger, ils activent des voies de signalisation qui finissent par converger pour induire la synthèse de cytokines pro-inflammatoires et d’interférons de type I. Les premières activent les cellules du système immunitaire tandis que les seconds participent notamment à la mise en place d’un système de défense antiviral.
Références
- G. Y. Chen and G. Nuñez, “Sterile inflammation : sensing and reacting to damage,” Nat. Rev. Immunol., vol. 10, no. 12, pp. 826–837, Dec. 2010.
- H. Kono and K. L. Rock, “How dying cells alert the immune system to danger,” Nat. Rev. Immunol., vol. 8, no. 4, pp. 279–289, Apr. 2008.
- P. Matzinger, “Tolerance, danger, and the extended family,” Annu. Rev. Immunol., vol. 12, pp. 991–1045, 1994.
- Y. J. Crow and N. Manel, “Aicardi-Goutières syndrome and the type I interferonopathies,” Nat. Rev. Immunol., vol. 15, no. 7, pp. 429–440, Jul. 2015.
- A. Härtlova et al., “DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity,” Immunity, vol. 42, no. 2, pp. 332–343, Feb. 2015.
- S. Akira, S. Uematsu, and O. Takeuchi, “Pathogen recognition and innate immunity,” Cell, vol. 124, no. 4, pp. 783–801, Feb. 2006.
- J. J. Chang and M. Altfeld, “TLR-mediated immune activation in HIV,” Blood, vol. 113, no. 2, pp. 269–270, Jan. 2009.
- J. M. Lund et al., “Recognition of single-stranded RNA viruses by Toll-like receptor 7,” Proc. Natl. Acad. Sci. U.S.A., vol. 101, no. 15, pp. 5598–5603, Apr. 2004.
- L. Alexopoulou, A. C. Holt, R. Medzhitov, and R. A. Flavell, “Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3,” Nature, vol. 413, no. 6857, pp. 732–738, Oct. 2001.
- H. Hemmi et al., “Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway,” Nat. Immunol., vol. 3, no. 2, pp. 196–200, Feb. 2002.
- O. Takeuchi and S. Akira, “Pattern recognition receptors and inflammation,” Cell, vol. 140, no. 6, pp. 805–820, Mar. 2010.
- S. N. Lester and K. Li, “Toll-like receptors in antiviral innate immunity,” Journal of molecular biology, vol. 426, no. 6, pp. 1246–64, Mar. 2014.
- A. Okumura, P. M. Pitha, A. Yoshimura, and R. N. Harty, “Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1,” J. Virol., vol. 84, no. 1, pp. 27–33, Jan. 2010.
- P. Georgel et al., “Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway,” Virology, vol. 362, no. 2, pp. 304–313, Jun. 2007.
- H. Kumar, T. Kawai, and S. Akira, “Toll-like receptors and innate immunity,” Biochem Biophys Res Commun, vol. 388, no. 4, pp. 621–5, Oct. 2009.
- D.-W. Yeh, L.-R. Huang, Y.-W. Chen, C.-Y. F. Huang, and T.-H. Chuang, “Interplay between Inflammation and Stemness in Cancer Cells : The Role of Toll-Like Receptor Signaling,” J Immunol Res, vol. 2016, p. 4368101, 2016.
- K. A. Fitzgerald et al., “IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway,” Nat. Immunol., vol. 4, no. 5, pp. 491–496, May 2003.
- M. Yamamoto et al., “Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway,” Science, vol. 301, no. 5633, pp. 640–643, Aug. 2003.
- E. Tomasello, E. Pollet, T.-P. Vu Manh, G. Uzé, and M. Dalod, “Harnessing Mechanistic Knowledge on Beneficial Versus Deleterious IFN-I Effects to Design Innovative Immunotherapies Targeting Cytokine Activity to Specific Cell Types,” Front Immunol, vol. 5, p. 526, 2014.
- D. Goubau, S. Deddouche, and E. S. C. Reis, “Cytosolic sensing of viruses,” Immunity, vol. 38, no. 5, pp. 855–69, May 2013.
- J. Wu and Z. J. Chen, “Innate immune sensing and signaling of cytosolic nucleic acids,” Annu. Rev. Immunol., vol. 32, pp. 461–488, 2014.
- M. Kaileh and R. Sen, “NF-κB function in B lymphocytes,” Immunol. Rev., vol. 246, no. 1, pp. 254–271, Mar. 2012.
- V. Hornung, “SnapShot : Nucleic acid immune sensors, part 2,” Immunity, vol. 41, no. 6, pp. 1066-1066.e1, Dec. 2014.
- S. W. Brubaker, K. S. Bonham, I. Zanoni, and J. C. Kagan, “Innate immune pattern recognition : a cell biological perspective,” Annu. Rev. Immunol., vol. 33, pp. 257–290, 2015.
- M. Yoneyama et al., “Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity,” J. Immunol., vol. 175, no. 5, pp. 2851–2858, Sep. 2005.
- H. Kato et al., “Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses,” Nature, vol. 441, no. 7089, pp. 101–5, May 2006.
- E. Meylan et al., “Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus,” Nature, vol. 437, no. 7062, pp. 1167–1172, Oct. 2005.
- S. Reikine, J. B. Nguyen, and Y. Modis, “Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5,” Frontiers in immunology, vol. 5, p. 342, 2014.
- L.-G. Xu, Y.-Y. Wang, K.-J. Han, L.-Y. Li, Z. Zhai, and H.-B. Shu, “VISA is an adapter protein required for virus-triggered IFN-beta signaling,” Mol. Cell, vol. 19, no. 6, pp. 727–740, Sep. 2005.
- H. S. Jin, H.-W. Suh, S.-J. Kim, and E.-K. Jo, “Mitochondrial Control of Innate Immunity and Inflammation,” Immune Netw, vol. 17, no. 2, pp. 77–88, Apr. 2017.
- H. Kato et al., “Cell type-specific involvement of RIG-I in antiviral response,” Immunity, vol. 23, no. 1, pp. 19–28, Jul. 2005.
- A. Dempsey and A. G. Bowie, “Innate immune recognition of DNA : A recent history,” Virology, vol. 479–480, pp. 146–152, May 2015.
- A. P. West et al., “Mitochondrial DNA stress primes the antiviral innate immune response,” Nature, vol. 520, no. 7548, pp. 553–557, Apr. 2015.
- L. Unterholzner, “The interferon response to intracellular DNA : Why so many receptors ?,” Immunobiology, Jul. 2013.
- T. Abe, Y. Marutani, and I. Shoji, “Cytosolic DNA-sensing immune response and viral infection,” Microbiol. Immunol., vol. 63, no. 2, pp. 51–64, Feb. 2019.
- K. Buchmann, “Evolution of Innate Immunity : Clues from Invertebrates via Fish to Mammals,” Front Immunol, vol. 5, p. 459, 2014.