L’édition génomique désigne la modification ciblée du matériel génétique d’un organisme vivant, procaryote ou eucaryote. Les mutations introduites peuvent concerner un à quelques nucléotides (substitutions, insertions, délétions) ou un gène entier qui peut être introduit (knock-in) ou inactivé (knock-out). Selon le but recherché, différents outils moléculaires sont employés. Cet article propose de passer en revue les principaux systèmes d’édition génomique, de leur fonctionnement moléculaire à leurs applications en recherche fondamentale et pratique, ainsi que leurs limites et les pistes de recherche pour les dépasser.
Fonctionnement moléculaire
Les systèmes de l’ère pré-CRISPR : méganucléases, nucléases à doigt de zinc et TALEN
En 1986, le laboratoire de Bernard Dujon, à l’Institut Pasteur, découvre la méganucléase I-SceI. Il s’agit d’une enzyme de restriction dont le site de reconnaissance présente une longueur inédite de 18 paires de bases 1, séquence si longue qu’elle n’existe pas dans les génomes eucaryotes. Au début des années 1990, des sites I-SceI sont introduits artificiellement dans le génome de plusieurs espèces, ce qui permet ensuite d’utiliser l’enzyme pour modifier le génome de celles-ci de manière précise et, surtout, avec une efficacité jusqu’à 1000 fois supérieure à celle les méthodes alors disponibles. La preuve était faite du potentiel des méganucléases pour l’édition du génome. Ces endonucléases reconnaissent en effet des sites particulièrement longs, d’au moins douze paires de bases, par rapport à la plupart des enzymes de restriction qui en reconnaissent six en moyenne. Sur une molécule d’ADN d’une taille donnée, les méganucléases vont donc engendrer moins de coupures que d’autres enzymes de restriction, ce qui permet des coupures plus spécifiques de l’ADN2. Il restait alors, pour être capable de cibler des séquences d’intérêt, le défi immense d’être capable de créer des méganucléases sur mesure 3.
Par la suite, des enzymes de restrictions artificielles plus versatiles sont créées en mettant bout à bout une séquence de nucléase non spécifique (domaine catalytique) avec une séquence de facteur de transcription (domaine de reconnaissance de l’ADN). Ces protéines de fusion appartiennent à deux grandes familles : les nucléases à doigts de zinc (zinc finger nucleases ou ZFN en anglais) et les TALEN4. La séquence ciblée par la nucléase dépend des domaines de reconnaissance des facteurs de transcription, qui sont choisis par l’expérimentateur 5.
Dans ces trois systèmes, la séquence cible d’ADN est reconnue uniquement par des protéines. Cibler une nouvelle séquence d’intérêt nécessite donc de trouver ou de mettre au point une protéine capable de s’y lier spécifiquement, un processus long et coûteux. Ces contraintes expliquent la révolution apportée par le système CRISPR/Cas9.
Nucléases et enzymes de restriction
Les nucléases sont des enzymes capables d’hydrolyser les liaisons entre nucléotides, c’est-à-dire de « couper » les acides nucléiques. Les désoxyribonucléases coupent l’ADN tandis que les ribonucléases coupent l’ARN. Si l’hydrolyse a lieu à l’extrémité de la chaîne de nucléotides, il s’agit d’exonucléases, si elle a lieu l’intérieur, il s’agit d’endonucléases.
Les enzymes de restriction sont des endodésoxyribonucléases. Elles reconnaissent des séquences spécifiques de l’ADN, appelées sites de restriction et coupent alors l’ADN au niveau de ce site ou à distance de ce site. Les enzymes de restriction capables de couper l’ADN au niveau du site de restriction sont très utilisées en génie génétique puisqu’elles permettent de cibler très précisément une séquence à couper.
La révolution CRISPR
Le système CRISPR, pour Clustered Regularly Interspaced Short Palindromic Repeats, est un mécanisme de défense des bactéries contre les infections virales1 2. Ce système immunitaire bactérien peut cependant être détourné et utilisé en biotechnologie pour cibler des séquences d’ADN ou d’ARN de manière précise 3.
Les systèmes d’éditions génomique dérivés de CRISPR/Cas reposent sur deux éléments essentiels : une molécule d’ARN guide (ARNg) et une endonucléase de la famille Cas (CRISPR associated protein) 4. L’ARNg est composé d’une séquence complémentaire à la séquence d’acide nucléique ciblée (d’une vingtaine de nucléotides) et d’une partie qui interagit avec la nucléase Cas (Figure 1). La famille Cas comporte de nombreuses protéines qui réalisent soit des coupures simple brin, soit des coupures double brin, et ciblent l’ADN ou l’ARN 56.
Une spécificité du système CRISPR/Cas est que l’endonucléase ne peut agir qu’à proximité d’une séquence appelée motif adjacent aux protoespaceurs (PAM en anglais, pour Protospacer Adjacent Motif) présente dans le génome cible 7. Le motif PAM est nécessaire pour la fixation de la Cas sur l’ADN : la protéine parcourt l’ADN et reconnaît les séquences PAM. Si la séquence en amont du PAM est complémentaire à l’ARN guide, alors la coupure de l’ADN a lieu. Ces séquences PAM sont spécifiques pour chaque membre de la famille Cas et comportent entre 2 et 6 nucléotides. Pour la protéine Cas9 de la bactérie Streptococcus pyogenes, la séquence PAM canonique est NGG, avec N qui peut être n’importe quel nucléotide, comme présenté en Figure 1.
Dans la suite, nous allons nous concentrer sur le système Cas9, le plus utilisé en édition génomique. Après liaison du complexe ARNg/Cas9 sur l’ADN cible, la coupure double brin a lieu 3 paires de bases en amont du site PAM. Cette coupure induit des mécanismes de réparation.
La cassure peut être réparée par un mécanisme de réparation non homologue (NHEJ pour Non Homologous End Joining), généralement imprécis, et qui provoque donc des insertions-délétions au niveau du site de coupure 8. Ceci permet d’obtenir l’inactivation d’un gène (encore appelée knock-out) en décalant le cadre de lecture, ce qui empêche la synthèse correcte de la protéine correspondante.
La coupure peut aussi être réparée par un mécanisme de recombinaison homologue dans le cas où une séquence exogène, dite matrice ou template, a été introduite en plus du complexe ARNg/Cas. Cette séquence peut alors être recopiée dans le génome de l’hôte 9. Pour cela, la séquence exogène doit comporter deux bras d’homologie qui permettent la recombinaison avec le génome de l’hôte. La séquence d’intérêt est située entre les deux bras d’homologies (Figure 1). Il peut s’agir d’un gène dans le cas d’un knock-in ou d’une mutation ponctuelle.
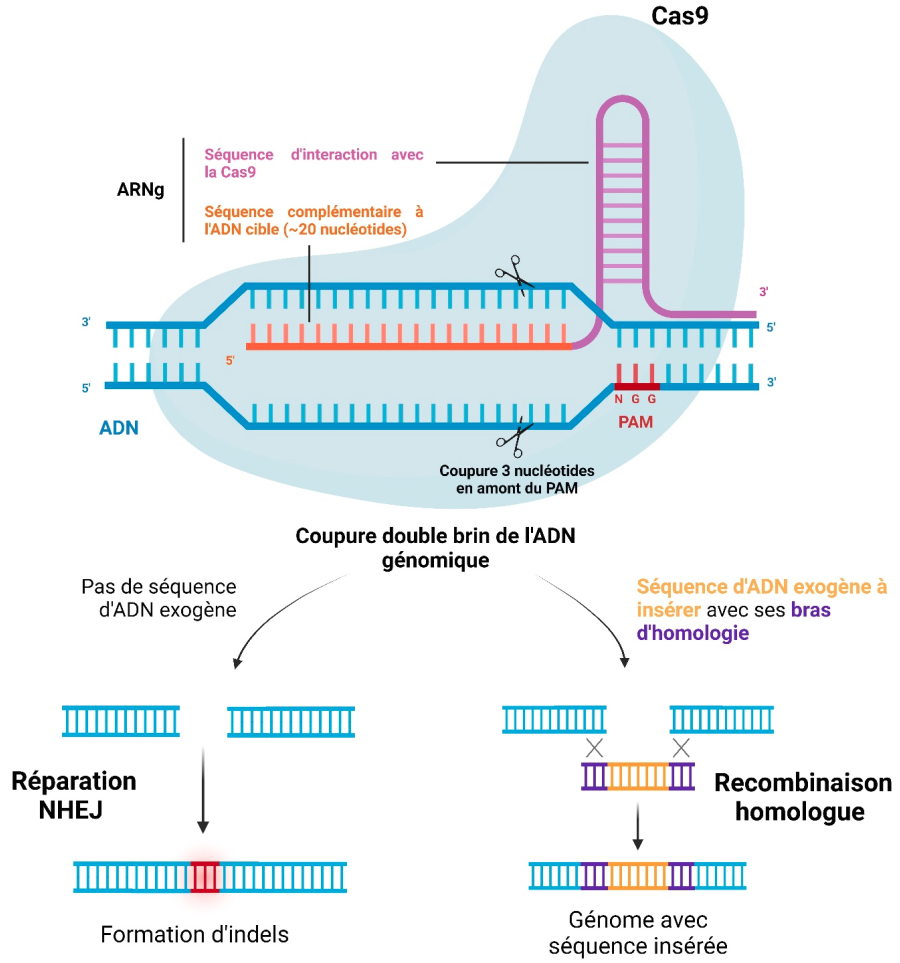
Détail du complexe Cas9/ARNg fixé sur sa séquence cible d’ADN. Après coupure double brin trois nucléotides en amont du motif PAM, deux mécanismes sont possibles. La réparation non homologue (NHEJ pour Non Homologous End Joining) induit des mutations aléatoires de type insertion ou délétion (indels). Si l’expérimentateur introduit une séquence avec des bras d’homologie, elle sera utilisée pour réparer la cassure et sera intégrée dans le génome par recombinaison homologue. Réalisé à l’aide de Biorender.
Après CRISPR : la réécriture par matrice d’ARN et la réécriture de base
Comme nous avons pu le voir, la modification par CRISPR/Cas permet d’altérer le génome de l’hôte de manière ciblée. Cette technologie est cependant souvent peu efficace lorsqu’il s’agit d’effectuer des modifications précises, car la réparation non homologue est bien plus fréquente que la réparation par recombinaison homologue. Deux systèmes d’édition génomique ont récemment émergé pour pallier ces lacunes : la réécriture par matrice d’ARN (prime editing) et la réécriture de base (base editing) 1.
La réécriture par matrice d’ARN (prime editing)
La réécriture par matrice d’ARN est un système permettant de réaliser de petites substitutions de nucléotides sans impliquer la voie de recombinaison homologue (Figure 2). Cette technologie utilise une protéine de fusion entre une Cas9 mutante, réalisant uniquement une coupure simple brin (Cas9 nickase ou nCas9), et une transcriptase inverse (RT, pour reverse transcriptase) permettant de copier la modification voulue au niveau de la séquence cible. Aucune matrice d’ADN n’est nécessaire pour réaliser la modification puisque l’ARN guide modifié, nommé ARNpeg (pour prime editing guide) est un ARN « trois-en-un », contenant les domaines suivants :
- une séquence guide permettant de cibler la coupure simple brin par nCas9-RT sur la séquence d’intérêt ;
- un site de liaison auquel l’ADN simple brin s’hybride (primer binding site, PBS) ;
- une matrice pour l’étape de transcription inverse.
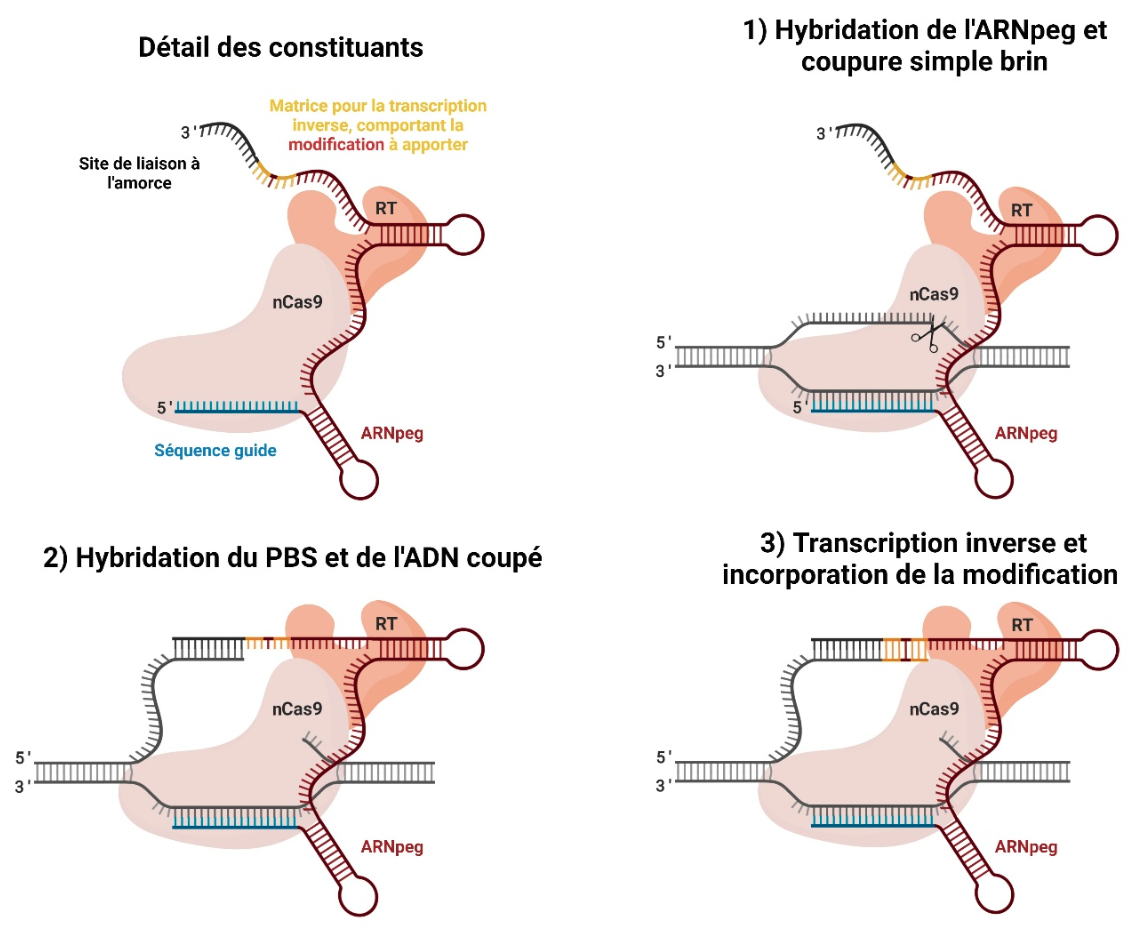
RT : transcriptase inverse (reverse transcriptase). PBS : site de liaison à l’amorce (primer binding site).
Étape 1 : Grâce à sa séquence cible, l’ARNpeg s’hybride à la séquence d’intérêt sur la molécule d’ADN ciblée. La nCas9 réalise alors une coupe simple brin.
Étape 2 : Le site de liaison à l’amorce (PBS) s’hybride avec la séquence complémentaire sur le brin d’ADN coupé.
Étape 3 : La transcriptase inverse prolonge le brin d’ADN coupé en polymérisant les nucléotides complémentaires de la séquence présente au niveau de l’ARNpeg. Cette séquence comporte la modification d’intérêt. La transcription inverse s’arrête généralement quand la transcriptase inverse bute sur la structure en épingle à cheveux de l’ARNpeg.
Après la transcription inverse, une étape de réparation des dommages simple brin permet de fixer la mutation dans le génome.
Le taux d’édition, c’est-à-dire le nombre de cellules ayant incorporé la modification souhaitée par rapport au nombre total de cellules qui ont été utilisées pour la manipulation, n’est cependant jamais de 100 %. Ce taux dépend de nombreux facteurs dont les plus influents sont la lignée cellulaire utilisée, la taille et le site de la modification. Ainsi, pour une modification de 5 paires de bases sur une lignée de cellules immortalisées, des taux d’édition de l’ordre de 20 % peuvent être atteints. En revanche, pour des inserts plus longs ou dans des cellules non-immortalisées et qui possèdent donc des systèmes de réparation des mésappariements fonctionnels, les taux d’éditions peuvent descendre en dessous de 0,1 % 123.
La réécriture de base (base editing)
La réécriture de base est quant à elle une technique qui permet la conversion ponctuelle de base de type C→T ainsi que A→G. Le mécanisme repose sur des enzymes modifiant les bases (cytosine désaminase par exemple) couplées avec une Cas9 nickase comme l’illustre la figure 3.
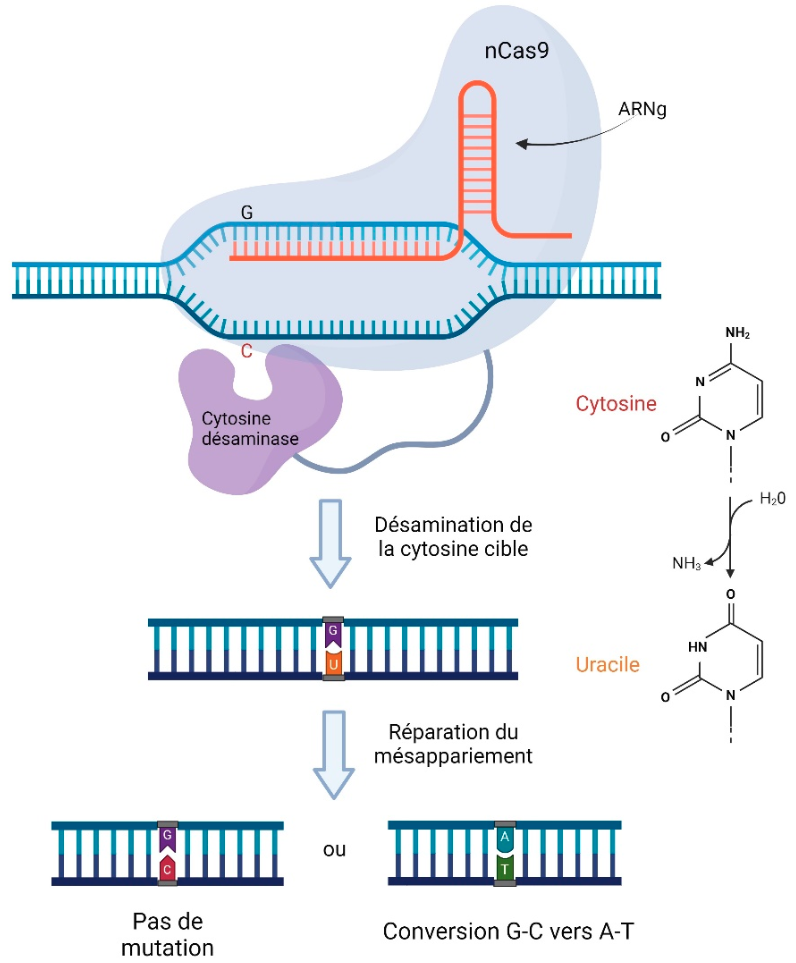
Après recrutement de la Cas9 fusion à la séquence cible, l’enzyme (ici, la cytosine désaminase) modifie chimiquement une base, conduisant à la modification voulue. Réalisé à l'aide de Biorender.
Dans l’étape de réparation du mésappariement, si le brin non modifié est utilisé comme matrice, l’uracile est remplacé par une cytosine, ce qui revient à la situation initiale. En revanche si le brin contenant l’uracile est utilisé comme matrice, cela aboutit à une paire U-A, qui est ensuite remplacée par une paire T-A au cours de la réplication.
Une des limites de cette technologie est que l’enzyme agit de manière aléatoire sur tous les nucléotides à sa portée, généralement dans une zone de cinq bases dans la partie 5’ du site d’hybridation de l’ARN guide. Une étape de sélection des cellules éditées est donc nécessaire pour conserver uniquement les modifications d’intérêt 1.
Les méthodes de transfert
Pour agir sur le génome, les complexes protéines/ARN que nous venons d’évoquer doivent d’abord entrer dans la cellule : une étape de transfert du matériel est donc nécessaire en amont.
De nombreuses méthodes de transfert ont été développées et sont utilisées selon les contextes cellulaires. Dans le cas des vecteurs viraux, une partie du génome viral est remplacée par une séquence exprimant la Cas9 et une autre exprimant l’ARN guide 2. Cette méthode est très efficace puisque les mécanismes de transfert du matériel génétique des virus ont été optimisés par l’évolution. Les virus adéno-associés figurent parmi les vecteurs viraux les plus employés, car ils sont peu immunogènes et ont la particularité de ne pas s’intégrer dans le génome de l’hôte, permettant une expression transitoire de la machinerie d’édition génomique 3.
Les méthodes non virales reposent sur des principes physiques ou chimiques. L’électroporation est une méthode très utilisée en laboratoire. Elle consiste à exposer brièvement les cellules à un choc électrique : cela crée transitoirement des pores dans la membrane plasmique, laissant passer des plasmides codant les protéines ou directement la protéine Cas associée à l’ARNg sous forme de ribonucléoprotéine 4. L’empaquetage des protéines et ARNg peut aussi se faire sous la forme de nanoparticules lipidiques, permettant la fusion avec la membrane de la cellule cible pour délivrer le matériel 5.
Le choix de la méthode de transfert dépend de nombreux critères, parmi lesquels la modification in vitro ou in vivo, la sensibilité des cellules et la taille du système d’édition. La figure 4 présente de manière visuelle les principales méthodes de transfert des systèmes d’édition génomique.
VAA : virus adéno-associés. Réalisé à l’aide de Biorender.
Édition génomique et recherches fondamentales
Par les possibilités qu’elles ouvrent en termes d’études de la fonction des gènes, les technologies d’édition du génome ont été une véritable révolution en recherche fondamentale. Avec ces outils, il est par exemple possible d’inactiver précisément une séquence cible pour en déduire son rôle physiologique, d’ajouter des gènes codant des protéines fluorescentes (par exemple la protéine fluorescente verte, GFP) afin de marquer aisément une population cellulaire ou encore de reproduire des mutations dans des cellules ou des organismes modèles pour étudier les pathologies associées.
Les systèmes d’édition génomique ont été un apport aussi bien pour les méthodes de génétique directe (forward genetics) que de génétique inverse (reverse genetics, Figure 5).
La génétique directe cherche à identifier des gènes à partir d’un phénotype donné : il s’agit de provoquer aléatoirement des mutations dans le génome d’individus à l’aide d’agents mutagènes physiques ou chimiques, puis de sélectionner les mutants correspondant au phénotype d’intérêt (Figure 5A). À partir de ceux-ci, il est possible de remonter au gène muté. Si cette technique de criblage génétique a été fondamentale dans l’histoire des sciences (par exemple pour caractériser les gènes impliqués dans le développement embryonnaire de la drosophile), elle est en revanche assez longue à mettre en place.
Les cribles génétiques ont été grandement facilités par l’outil CRISPR. En utilisant des banques d’ARNg, il est possible d’inactiver l’ensemble des gènes, sans à priori sur des gènes candidats 1. Les ARNg sont transférés aléatoirement dans les cellules étudiées mais leurs séquences sont connues : ainsi, en cas de phénotype intéressant, il est possible de remonter à la séquence qui a été mutée en réalisant un simple alignement de séquence entre l’ARNg et le génome de référence de l’organisme modèle. L’étape d’identification de la séquence mutée est donc beaucoup plus simple que dans les techniques de mutagenèse aléatoire plus anciennes.
La génétique inverse prend le parti opposé de la génétique directe : il s’agit de muter spécifiquement un gène candidat et d’observer les modifications phénotypiques afin de déduire sa fonction (Figure 5B). Cette approche requiert deux présupposés : connaître la séquence que l’on veut muter et être capable de cibler spécifiquement cette séquence, ce dernier point étant rendu possible par les systèmes d’édition génomique.
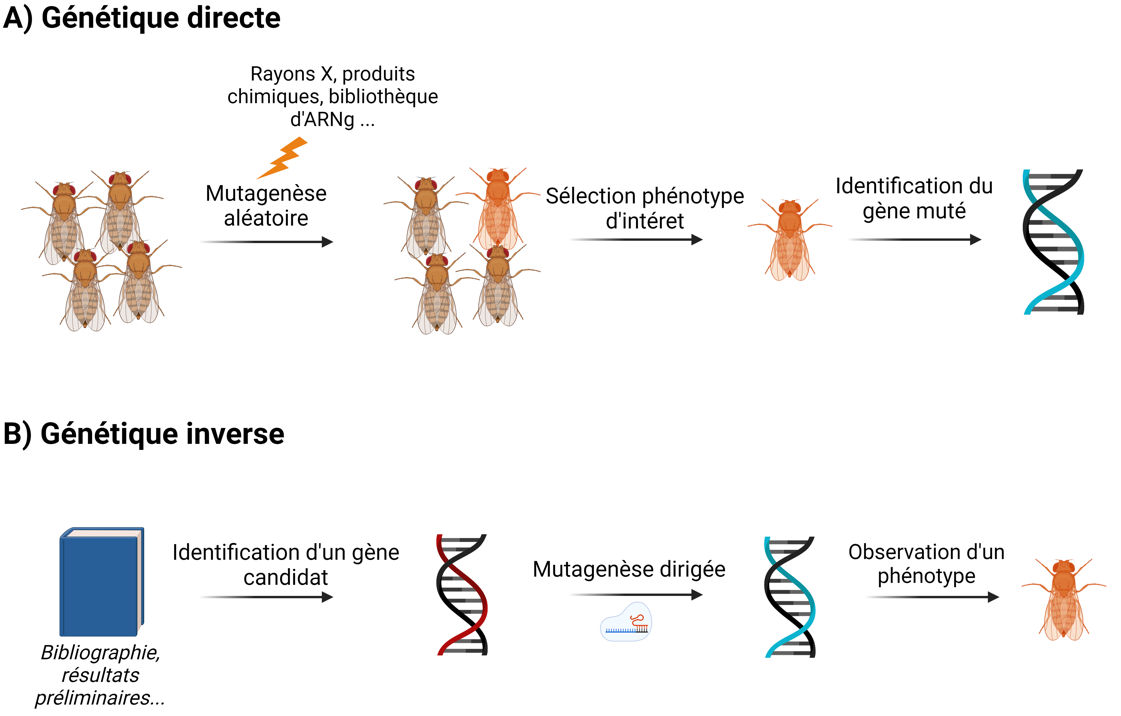
En génétique directe (A), on part sans à priori et l’on déduit le gène impliqué dans une fonction grâce au phénotype observé sur un mutant d’intérêt. En génétique inverse (B), on sélectionne préalablement un candidat à l’aide de données préliminaires, que l’on mute spécifiquement pour observer ensuite les conséquences de sa délétion. Réalisé à l’aide de Biorender.
Des applications variées en agronomie et médecine
En agronomie
Les systèmes d’édition génomique possèdent de nombreuses applications dans le domaine agronomique, notamment pour la production d’organismes génétiquement modifiés (OGM). En Europe, les OGM sont définis comme tout organisme dont le patrimoine génétique a été modifié grâce au génie génétique. L’introduction de mutations permet ainsi de propager de nouveaux allèles dans les populations et d’obtenir des plantes ou animaux ayant les caractères recherchés : fruits de plus grosse taille, résistance à la sécheresse ou aux pathogènes, hypertrophie musculaire du bétail… 1
Si la modification génétique des plantes est appliquée depuis des millénaires (sélection artificielle des variétés les plus faciles à cultiver par exemple), l’édition génomique avec le système CRISPR/Cas permet d’accélérer ces processus et de contrôler finement le génome des plantes cibles. L’addition de nouveaux traits est possible depuis plusieurs dizaines d’années, grâce au transfert horizontal de gènes par la bactérie Agrobacterium tumefaciens 2, mais est maintenant remplacée par l’outil CRISPR. Les résultats de telles manipulations sont déjà sur le marché : en 2022, une race de bœuf résistante à la chaleur, obtenue par CRISPR/Cas9, a été autorisée à la consommation aux États-Unis 3.
En termes légaux, les régulations concernant l’édition du génome en agronomie varient selon les pays considérés. Dans l’Union européenne (qui possède la juridiction la plus stricte sur le sujet), les plantes transgéniques obtenues par CRISPR/Cas9 ou dérivées sont soumises aux mêmes réglementations que les produits OGM 4.
Outre-Atlantique, les mesures sont souvent plus souples. Le département de l’Agriculture des États-Unis (USDA), tout comme le Comité de biosécurité brésilien, ont ainsi décidé de ne pas ajouter de régulations spécifiques sur les plantes issues de l’édition génomique qui ne contiennent pas d’ADN étranger. L’argument majeur est que les modifications à l’échelle de quelques nucléotides s’apparentent à des mutations qui pourraient arriver naturellement. Ces plantes sont ainsi soumises à la même législation que les autres cultures 56.
En santé humaine
L’engouement autour de CRISPR s’explique notamment par les promesses apportées dans le domaine biomédical, en particulier dans le cadre des thérapies géniques 78. Celles-ci visent à soigner des maladies en remplaçant ou complémentant un allèle mutant défectueux par un allèle fonctionnel. De nombreuses pathologies pourraient théoriquement être soignées par cette méthode : mucoviscidose, maladies auto-immunes, myopathies…

Le principe de la thérapie génique est préexistant aux outils actuels d’édition génomique. En 2000, le premier succès d’une thérapie génique est annoncé après que les docteurs Alain Fischer, Marina Cavazzana-Calvo et Salima Hacein-Bey-Abina ont soignés des enfants atteints d’immunodéficience sévère (« bébés bulles »). Cependant, la méthode utilisait des vecteurs viraux pour apporter le gène fonctionnel et l’intégration aléatoire de la cassette dans le génome humain a été à l’origine de deux cas de leucémies sur les dix enfants de la cohorte 1. Cet événement freina temporairement les essais de thérapie génique en mettant l’accent sur la nécessité de sécuriser les méthodes.
Ces risques sont grandement diminués avec le système CRISPR/Cas9 puisque la modification est ciblée sur une séquence précise : il est possible de choisir la localisation de la cassette intégrée de manière à éviter les dérégulations de l’expression génomique globale. Les systèmes de réécriture par matrice d’ARN et de réécriture de base sont eux aussi prometteurs puisqu’ils permettraient de corriger des mutations ponctuelles, dans le cas de la mucoviscidose par exemple.
Détaillons maintenant les différentes étapes d’une thérapie génique. Après identification du gène défectueux chez le patient, deux situations peuvent se présenter suivant le tissu atteint. Si le tissu est accessible aux techniques de transfert existantes, les cellules du patient sont modifiées directement dans le corps du patient. C’est par exemple le cas du traitement de l’amaurose congénitale de Leber, une maladie de la rétine pour laquelle le traitement (Luxturna) est injecté directement dans les yeux du patient 2. Lorsque les cellules cibles ne sont pas directement accessibles, la thérapie nécessite de prélever les cellules du patient puis de réaliser la transformation des cellules in vitro, de sélectionner les cellules transgéniques souhaitées et de les réinjecter dans le patient. Cette méthode est par exemple utilisée dans le traitement des maladies auto-immunes ou encore de la drépanocytose, illustrée en figure 6, pour laquelle un traitement (Casgevy®, par Vertex Pharmaceuticals) a reçu l’autorisation de mise sur le marché britannique en novembre 2023 3.
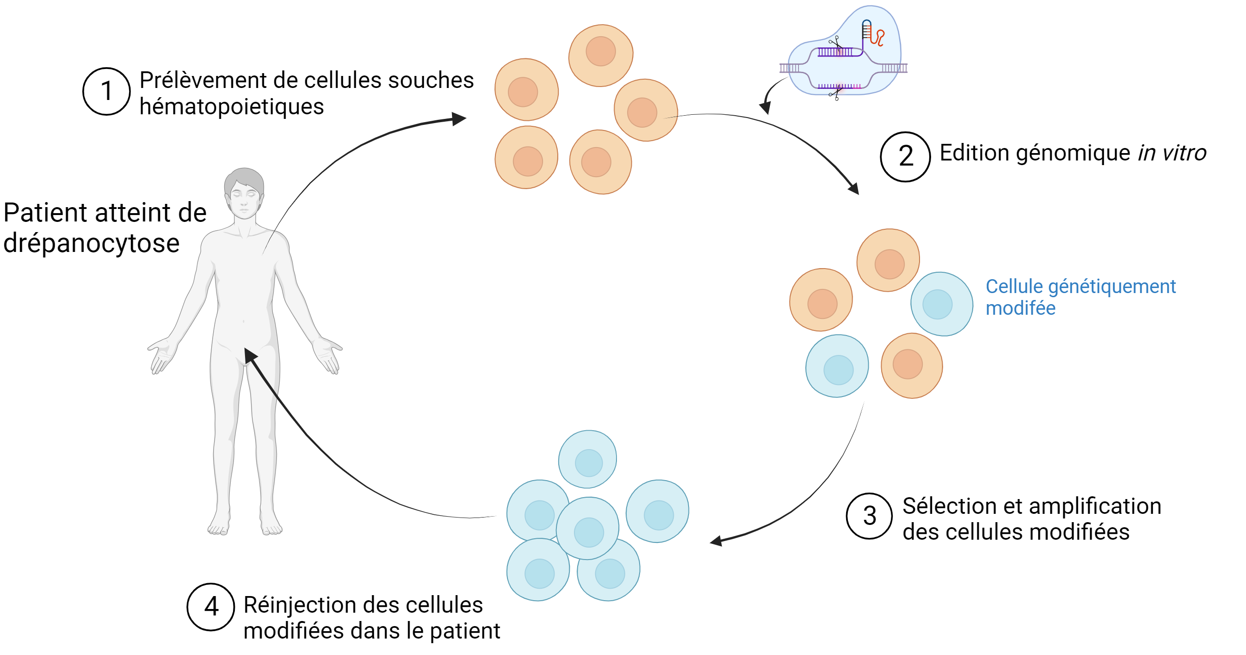
Dans le cas de la drépanocytose, l’édition génomique est faite in vitro après prélèvement des cellules souches hématopoïétiques (précurseurs des globules rouges) du patient. Réalisé à l’aide de Biorender.
Ces possibilités de modifications du génome humain posent des questionnements éthiques 12. Rappelons qu’en France, comme dans les autres pays européens ayant adopté la convention d’Oviedo 3, seule la modification du génome des cellules somatiques est autorisée chez l’humain, l’édition des gamètes étant prohibée 4.
Les limites de l’édition génomique
Efficacité et spécificité
Une des premières limites techniques réside dans l’efficacité des manipulations d’édition génomique précise. En effet, l’insertion d’une séquence exogène est un phénomène relativement rare puisqu’il nécessite une réparation par recombinaison homologue dont l’activité est généralement moins forte que la réparation non homologue (NHEJ). L’étape d’isolement et de sélection des mutants est donc à prendre en compte dans la durée et les coûts de la manipulation. Pour des applications en recherche, la sélection des cellules mutantes peut s’effectuer en insérant, en plus de la séquence d’intérêt, un gène codant une molécule fluorescente ou un gène de résistance à un antibiotique.
Un second problème inhérent à l’édition génomique est celui de sa spécificité. En effet, l’ARNg est conçu pour se lier spécifiquement à une séquence d’ADN d’intérêt par complémentarité des bases, mais il peut aussi se lier à des séquences homologues proches (à quelques nucléotides près) 5. Les recherches actuelles cherchent à diminuer l’occurrence de ces évènements pour sécuriser au maximum l’édition génomique et éviter des altérations non désirées potentiellement délétères dans le cas d'applications thérapeutiques.
Stratégies de transfert
Un aspect technique évoqué précédemment est celui du transfert des systèmes d’édition. Si les vecteurs viraux permettent de faire exprimer efficacement des protéines par la cellule infectée, il reste le problème de la taille des inserts.
En effet, les virus adéno-associés possèdent un génome de seulement 4700 paires de bases. Il est ainsi difficile de faire rentrer dans le même vecteur la séquence codante pour la protéine Cas9 et une cassette d’ADN exogène. La capacité limitée des virus adéno-associés restreint donc les possibilités d’utilisation de cette méthode de transfert pour l’édition génomique 6.
Pour résoudre ces problématiques, de nombreuses approches sont poursuivies : utilisation d’autres vecteurs viraux (qui posent alors des problèmes de réponse immunitaire de l’hôte) ou encore miniaturisation des systèmes (mini-Cas9). Le Tableau 1 récapitule les méthodes utilisées, leurs avantages et inconvénients.
| Méthode | Principe | Avantages | Inconvénients |
|---|---|---|---|
| Vecteur viral | Insérer une séquence d’expression de la protéine Cas et de l’ARNg dans le génome viral, puis infecter les cellules cibles avec ces virus modifiés | — Infection efficace — Expression transitoire car pas d’intégration du virus dans le génome hôte — Usage in vivo et in vitro |
— Immunogénicité potentielle des virus — Taille de l’insert limitée |
| Micro-injection | Injecter directement le matériel dans la cellule à l’aide de fines aiguilles | — Acheminement direct sans intermédiaire — Usage in vivo et in vitro |
Limités à certains systèmes biologiques (cellule-œuf) |
| Électroporation | Appliquer un champ électrique pour créer des pores temporaires dans la membrane | — Permet d’introduire directement la protéine, sans étape d’expression d’un plasmide | — Certains types cellulaires peuvent être trop sensibles pour l’électroporation — Ne peut être utilisé in vivo |
| Nanoparticules lipidiques | Encapsuler les protéines et l’ADN chargé négativement dans des liposomes chargés positivement | — Peut être utilisé in vitro ou in vivo — Les cargos sont protégés des protéases extracellulaires |
Dégradation possible des endosomes après internalisation |
Limites médicales et coûts associés
Si de nombreux espoirs reposent sur la thérapie génique pour soigner des pathologies encore incurables, soulignons que nous en sommes encore aux balbutiements. Peu d’essais cliniques ont été portés à leurs termes, et le nombre de cibles est relativement faible. Pour l’instant seules les maladies monogéniques sont traitées, c’est-à-dire celles causées par un dysfonctionnement d’un unique gène connu : le traitement de maladies multigéniques demande le développement de nouvelles techniques et des protocoles thérapeutiques associés 7. De plus, les thérapies se concentrent sur un unique tissu : muscles dans les dystrophies de Duchenne, rétine pour l’amaurose de Leber, moelle osseuse pour certaines immunodéficiences 89.
Enfin, il ne faut pas oublier que ces thérapies révolutionnaires sont accompagnées d’un coût qui limite grandement leurs applications à grande échelle. En 2022, l’Agence fédérale américaine des produits alimentaires et médicamenteux (FDA) a ainsi autorisé la mise sur le marché d’une thérapie génique pour traiter l’hémophilie de type B pour la somme de 3,5 millions de dollars par dose. Ce médicament, appelé Hemgenix, est devenu le plus cher du monde 1011.
De même, la société Vertex Pharmaceuticals a annoncé en 2023 une thérapie génique contre la drépanocytose d'un coût estimé entre 1 et 3 millions de dollars. Cela pose la question de l’accessibilité du traitement, lorsque l’on sait qu’en Afrique subsaharienne, zone géographique de prévalence majeure de la pathologie, les revenus annuels moyens étaient inférieurs à 2000 dollars en 2020 12.
Conclusion
Depuis la découverte des outils d’édition génomique à la fin des années 1990, les chercheurs disposent aujourd’hui d’outils efficaces pour provoquer des mutations, inactiver des gènes ou encore ajouter des séquences codantes d’ADN, et ce de manière spécifique. Ces systèmes d’édition n’ont cessé de se perfectionner, mais aussi de se diversifier : CRISPR/Cas9 reste ainsi crucial pour insérer ou remplacer de longs segments d’ADN, tandis que la réécriture par matrice d’ARN et la réécriture de base permettent des modifications plus précises, à l’échelle de quelques nucléotides, pour corriger une mutation ponctuelle. Enfin, si les systèmes CRISPR/Cas sont majoritairement utilisés pour l’édition génique, la modification de l’épigénome a récemment pris de l’ampleur. Ainsi un mutant de Cas9 dépourvu d’activité de coupure, fusionné à d’autres domaines enzymatiques, permet d’induire des modifications épigénétiques de manière spécifique 13. Malgré toutes ces promesses, tant dans les domaines fondamentaux que pratiques, ces technologies sont récentes et continuent d’être améliorées.
Enfin, les possibilités considérables apportées par ces outils posent de nouveaux questionnements éthiques, ce qui peut amener à repréciser l’encadrement réglementaire existant afin d’éviter tout débordement. En 2018, l’annonce d’enfants nés suite à une modification de leur génome par CRISPR/Cas9 (« bébés CRISPR ») par le scientifique chinois He Jiankui avait provoqué une vague d’indignation au sein de la communauté scientifique 14. Cet exemple extrême aura rappelé la nécessité de mettre en place des régulations internationales strictes concernant l’édition génomique, tout en mettant en exergue la nécessité de dialogue entre scientifiques, dirigeants et grand public pour naviguer dans un paysage éthique oscillant au gré des nouvelles technologies.