Les pathologies dégénératives de la rétine représentent en Europe et dans le monde une cause importante d’altération de la vision et de cécité, et la cause majeure dans les pays à fort revenus. Certaines de ces pathologies sont héréditaires, car d’origine génétique, parmi lesquelles les rétinites pigmentaires, modèles de choix pour le développement de nombreuses pistes thérapeutiques. Les approches de thérapie génique pour le traitement de ces pathologies génétiques sont considérées comme les plus prometteuses, certaines d’entre elles ayant déjà atteint le stade d’études cliniques chez les patients. Plusieurs stratégies ont été développées, pouvant aller d’une modification du génome ne changeant que quelques paires de bases de l’ADN des cellules visées, à des stratégies beaucoup plus drastiques lorsque les cellules photosensibles ont déjà dégénéré. Un autre type cellulaire est alors détourné pour jouer un rôle de cellule photosensible alors qu’il ne possédait pas cette fonction initialement.
Les principales causes de déficiences visuelles à l’échelle mondiale
La perte sensorielle qui affecte le plus l’autonomie des personnes dans nos sociétés est la perte de la vue. Les principales causes de déficience visuelle à l’échelle mondiale sont 1 :
-
les erreurs de réfraction, dans lesquelles l’œil présente une irrégularité de forme qui provoque des défauts de vision. C’est le cas des problèmes de vision que la plupart d’entre nous connaissons le mieux, comme la myopie, l’hypermétropie ou encore l’astigmatisme. Ces erreurs peuvent être corrigées par le port de lunettes ou de lentilles.
-
la cataracte, qui est due à une opacification du cristallin, généralement causée par le vieillissement. Ce tissu fonctionne comme une lentille et permet la convergence des rayons lumineux sur la rétine. Son remplacement par une prothèse artificielle permet de restaurer la vision.
-
la rétinopathie diabétique, due à une altération des vaisseaux sanguins irriguant la rétine chez les personnes diabétiques. En effet, l’excès de glucose dans le sang peut provoquer des hémorragies, la formation de nouveaux vaisseaux anormaux (angiogenèse) ou encore des morts cellulaires au sein de la rétine. Ces différents phénomènes dégradent la vue des patients.
-
les maladies liées à une dégénérescence des cellules nerveuses de l’œil. L’exemple le plus connu du grand public est la dégénérescence maculaire liée à l’âge ou DMLA. Cette pathologie est liée au vieillissement et se traduit par une dégénérescence de cellules de la macula, la partie centrale de la rétine, provoquant la perte du centre du champ de vision des patients. D’autres formes de dégénérescence existent, comme les rétinites pigmentaires.
Les deux principales causes de déficiences visuelles à l’échelle mondiale (erreurs de réfraction et cataracte) peuvent être traitées ou compensées, et sont handicapantes majoritairement dans les pays à faibles revenus 2. Les maladies se caractérisant par une dégénérescence de cellules de la rétine constituent quant à elle l’objet d’un grand nombre de recherches, les options de traitements restant très limitées.
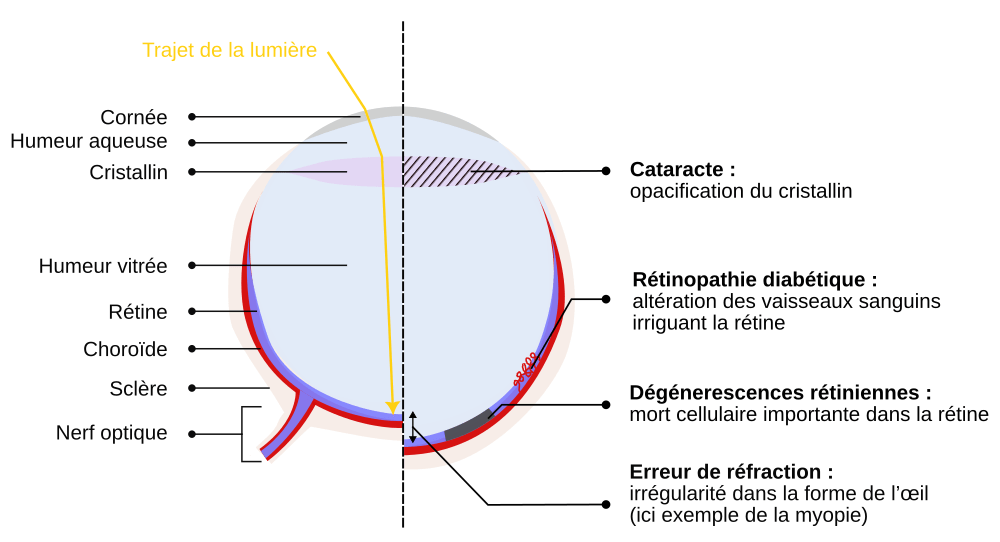
L’œil humain est délimité par deux tissus : la cornée, transparente et recouvrant une majorité de la zone visible de l’œil, et la sclère (ou sclérotique), blanche et résistante aux chocs, située sur le reste du globe oculaire. La lumière entre dans l’œil par la cornée, traverse un premier gel transparent, l’humeur aqueuse, puis arrive au cristallin. Le cristallin agit comme une lentille déviant les rayons lumineux et permettant leur convergence sur la rétine. La lumière traverse ensuite un deuxième gel transparent occupant la majeure partie de l’intérieur de l’œil, l’humeur vitrée et arrive au sein du tissu nerveux recouvrant le fond de la chambre oculaire : la rétine. Celle-ci contient des cellules photosensibles : cônes et bâtonnets, ainsi qu’un réseau de neurones qui permettra la transduction du signal lumineux en un signal électrophysiologique transféré au cerveau par le nerf optique. La choroïde, sous-jacente à la rétine, est constituée d’un réseau de vaisseaux sanguins et permet son approvisionnement en nutriments et gaz respiratoires.
Les dégénérescences rétiniennes et leur modèle expérimental privilégié : la rétinite pigmentaire
Les dégénérescences de la rétine représentent un groupe hétérogène de pathologies, dont la gravité, le déroulé et l’âge d’apparition dépendent de facteurs génétiques et environnementaux divers. Dans ce groupe de maladies, ce sont le plus fréquemment les photorécepteurs (les cônes et les bâtonnets, c’est-à-dire les cellules photosensibles de la rétine) qui finissent par dégénérer et causer la perte de vision.
Ces morts cellulaires peuvent être causées par l’âge et le vieillissement des tissus, comme c’est le cas dans la DMLA, ou être héréditaires, comme dans les rétinites pigmentaires. De nombreux travaux de recherche ont porté sur ces dernières car l’identification des mutations à leur origine permet d’imaginer des traitements correctifs du génome.
Les rétinites pigmentaires forment un ensemble de maladies héréditaires se traduisant par une perte progressive de la vision (Figure 2) 1 :
-
À l’adolescence, la vision de nuit se dégrade petit à petit, ne permettant plus de voir correctement en conditions de faible luminosité (stade 1 de la pathologie).
-
Avec l’âge apparaissent des troubles de la vision de jour, atteignant en premier la vision périphérique. Les personnes atteintes ne voient plus qu’au centre de leur champ de vision, développant une vision « tunnel ». La perte du champ de vision est d’autant plus centrale et marquée que l’âge avance (stade 2).
-
Entre 50 et 80 ans selon les patients, arrive une perte de la vision centrale, dernière à être atteinte (stades 3 et 4 : au stade 3 plus aucun photorécepteur n’est fonctionnel, au quatrième stade la dégénérescence des photorécepteurs est complète et ces cellules ont définitivement disparu de la rétine). La perte de vision devient totale.
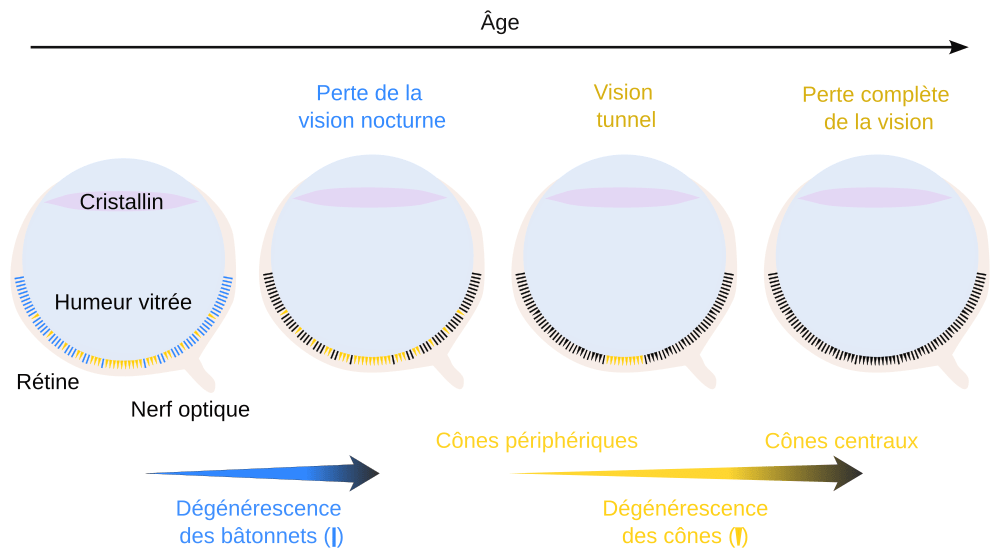
La perte de la vision de nuit, puis de la vision de jour, sont provoquées par une dégénérescence en premier lieu des bâtonnets (en bleu), suivi d’une dégénérescence des cônes (en jaune). Ces derniers dégénèrent de la périphérie vers le centre. Il a en effet été découvert que les bâtonnets produisaient des facteurs trophiques (RdCVF : rod-derived cone viability factor) nécessaires à la survie des cônes 1.
Pour comprendre l’origine de ces dégénérescences héréditaires, il a fallu commencer par identifier des gènes et des mutations responsables. Le gène de la rhodopsine a été l’un des premiers mis en cause dans la rétinite pigmentaire dans les années 90 1.
La rhodopsine est une protéine exprimée par les bâtonnets dans la rétine. Elle appartient à la famille des opsines, protéines permettant la sensibilité d’une cellule à la lumière. Les opsines possèdent deux composants principaux : une chaîne d’acides aminés et une molécule de rétinal, une des formes de la vitamine A. Le rétinal a une fonction de détecteur. Lorsqu’il absorbe un photon, il change de forme. Ce changement de forme va à son tour déformer la chaîne d’acides aminés de l’opsine, et permettre à la protéine de déclencher un message dans la cellule qui l’exprime, cône ou bâtonnet. Dans le cas de la mutation du gène de la rhodopsine à l’origine d’une des formes de rétinite pigmentaire, l’hypothèse expliquant la dégénérescence des bâtonnets est celle d’une atteinte de la séquence permettant la translocation de la rhodopsine dans la partie photosensible de la cellule, zone où elle est normalement exprimée. Ce défaut de translocation provoquerait à moyen terme la dégénérescence des cellules en engorgeant le système d’adressage des protéines, ou encore en interférant avec les autres processus cellulaires 2.
De nombreuses recherches ont depuis démontré que d’autres mutations que celles du gène de la rhodospine, en particulier des mutations touchant des gènes codant des protéines de la cascade de transduction du signal lumineux (comme RPE65 par exemple), pouvaient aboutir à des rétinites pigmentaires. Ces protéines, produites en très grandes quantités dans tous les cônes et les bâtonnets, peuvent s’accumuler et engorger la machinerie cellulaire de production et de recyclage des protéines lorsqu’elles sont non fonctionnelles, menant à une mort prématurée des cellules.
Les pistes thérapeutiques des dégénérescences rétiniennes
Les trois grandes pistes actuelles pour préserver ou restaurer la vue chez les patients atteints de dégénérescences rétiniennes sont :
-
Les implants rétiniens. Il s’agit de grilles d’électrodes remplaçant les cellules photoréceptrices : une fois activées par la lumière, elles stimulent directement les neurones de la rétine.
-
La thérapie régénérative, qui cherche à implanter de nouvelles cellules développées en culture à partir de cellules souches pour remplacer les cellules en dégénérescence.
-
La thérapie génique, actuellement la piste de traitement majeure, qui peut être définie dans son acception la plus large comme l’utilisation d’ADN exogène pour traiter une pathologie. Elle est envisagée comme traitement depuis les années 1970 3. Elle présente des avantages thérapeutiques évidents : un unique traitement pourrait provoquer un effet clinique curatif (et pas uniquement symptomatique) et pérenne.

Différentes stratégies de thérapie génique pour les rétinites pigmentaires
En théorie, le principe de la thérapie génique est simple : il s’agit de réparer une séquence d’ADN. En pratique, sa mise à en œuvre pose de nombreux défis.
Comment insérer un ADN exogène dans une cellule ?
Les cellules du corps humain possèdent des mécanismes évitant l’entrée ou encore l’insertion dans leur génome de molécules d’ADN d’origine exogène. Ces mécanismes sont précieux et nécessaires à la préservation du génome de nos cellules. Les premières techniques de thérapie génique proposées nécessitaient d’extraire des cellules des patients, de réaliser les protocoles de modification des cellules en laboratoire, puis de réimplanter ces cellules chez les patients.
De nombreuses découvertes dans le domaine de la virologie ont depuis révolutionné les outils à disposition permettant de contourner ces mécanismes. Certains virus sont en effet capables d’insérer une séquence d’ADN au sein du génome de leur cellule hôte. Dans le cadre de la thérapie génique, les virus adénoassociés (AAV adeno-associated virus en anglais) sont particulièrement intéressants. Ces petits virus à ADN, non pathogènes, peuvent infecter les cellules de primates, dont les cellules humaines. Ils sont appelés virus adénoassociés car leur réplication ne peut se faire qu’en cas de co-infection par certains autres virus, dont les adénovirus.

Comment cibler les cellules souhaitées uniquement ?
Depuis la découverte de ces virus adénoassociés, l’ingénierie biotechnologique a permis leur modification au point de pouvoir produire des virus capables d’infecter des cellules cibles et d’y introduire une séquence d’intérêt en provoquant peu à pas de réponse immunitaire ou inflammatoire. Ces virus sont nommés virus adénoassociés recombinants : leur matériel génétique initial est remplacé par une séquence choisie. Des modifications artificielles des protéines de leur capside ont même permis de produire des virus infectant de façon spécifique certains types cellulaires comme les photorécepteurs, permettant de diminuer fortement les risques que le traitement n’ait des conséquences sur d’autres cellules.
Selon la mutation, quelle séquence d’ADN insérer pour restaurer le fonctionnement du système ?
Une fois ces vecteurs développés, la question devient alors de savoir quelle séquence insérer à l’intérieur de leur capside pour compenser le gène muté.
Les premières approches imaginées, dont certaines ont actuellement passé le stade des premiers essais cliniques chez l’humain, consistent à ajouter dans la cellule un gène fonctionnel pour remplacer le gène muté. Cette stratégie est par exemple employée pour traiter des dystrophies rétiniennes héréditaires appartenant aux amauroses congénitales de Leber, liées à des mutations du gène RPE65. Dans ce cas, le traitement, appelé Luxturna, correspond à un vecteur viral adénoassocié, contenant une copie fonctionnelle du gène RPE65.
La stratégie simple d’ajout d’une copie fonctionnelle du gène muté, qui peut fonctionner lorsqu’il s’agit de pallier la fonction d’un gène muté ne produisant pas de protéine ou une protéine inactive, n’est malheureusement pas adaptée à toutes les situations. C’est notamment le cas des pathologies causées par une mutation menant à la formation de protéines toxiques. Apporter un gène non muté en supplément ne résout pas le problème de la présence du gène muté qui continue de provoquer la dégénérescence.
Dans ce cas de figure, il est possible de charger les virus adénoassociés avec des outils dérivés des ciseaux à ADN de type CRISPR/Cas9. Par exemple, les systèmes de réécriture de base (base editing en anglais) permettent d’utiliser les capacités de reconnaissance de séquence des systèmes CRISPR/Cas9 pour modifier spécifiquement une unique base dans la séquence du génome, et ainsi corriger une mutation délétère. Les virus adénoassociés recombinants servent alors de vecteurs à ce système de correction, en permettant l’infection des seules cellules visées (Figure 3).

Dans l’exemple présenté, un virus adéno-associé recombinant infectant spécifiquement les photorécepteurs est injecté au sein de la rétine. Ce virus contient une séquence codant deux éléments majeurs : la séquence codant la nucléase Cas9 (vert foncé) et la séquence d’un ARN guide (vert clair) spécialement conçu pour reconnaître la cible sur l’ADN à modifier (gène muté représenté en rouge). L’expression de ces séquences dans la cellule cible permet la correction du gène d’intérêt.
Comment adapter les stratégies au stade de la maladie ?
Les stratégies de thérapie génique doivent non seulement être adaptées aux différentes mutations mais aussi au stade auquel est traitée la pathologie : les cellules cibles idéales peuvent avoir déjà entièrement dégénéré, nécessitant un changement d’approche.
Stade 1
Si la pathologie est prise en charge au tout début de la dégénérescence des bâtonnets, les stratégies de thérapies géniques peuvent se concentrer sur le remplacement ou la compensation du gène muté (comme décrit précédemment).
Stades 2 et 3
Une fois le nombre de bâtonnets déjà drastiquement diminué par la dégénérescence, remplacer les gènes à l’origine de ces dégénérescences n’est plus une stratégie utilisable. Les thérapies actuellement en développement se concentrent alors sur l’augmentation de l’expression de facteurs trophiques sécrétés par les bâtonnets et nécessaires au maintien des cônes, les RdCVF (rod-derived cone viability factors). Il s’agit d’éviter le cercle vicieux où la dégénérescence des bâtonnets provoque celle des cônes.
Des thérapies géniques actuellement en développement pour des applications cliniques visent ainsi à faire exprimer RdCVF dans d’autres cellules que les bâtonnets.
Stade 4
Quelle stratégie envisager lorsque l’ensemble des photorécepteurs, cônes et bâtonnets ont déjà fini de dégénérer ? Remplacer le gène atteint, ou augmenter la concentration en facteurs trophiques de survie des photorécepteurs est alors inutile. Les chercheurs ont donc imaginé d’utiliser la thérapie génique pour rendre photosensibles des cellules qui ne l’étaient pas jusqu’ici. L’objectif est de restaurer la photosensibilité de la rétine en faisant exprimer une opsine dans une cellule du réseau neuronal de la rétine, cellule bipolaire ou cellule ganglionnaire. Cette stratégie originale s’appelle l’optogénétique.
Les premières thérapies développées sur ce schéma utilisent une opsine bactérienne, la rhodopsine canal (ou channelrhodopsine) ChrimsonR. Son fonctionnement est très simple, car ne nécessitant pas de cascade de transduction. Il s’agit en effet d’un canal ionique photosensible : sa stimulation par l’absorption d’un photon permet immédiatement une dépolarisation de la cellule.
ChrimsonR n’est cependant pas aussi sensible à la lumière que la rhodopsine endogène, notamment à cause de l’absence de cascade de transduction amplifiant le signal. Les essais cliniques réalisés chez l’humain utilisent donc un système de lunettes amplificatrices qui enregistrent la scène entourant le patient, et la projette sur la rétine des patients dans les longueurs d’onde optimales à la stimulation de ChrimsonR et avec une intensité plus importante que l’intensité de la lumière naturelle 1. Le système est similaire à un appareillage auditif qui enregistre les sons de l’environnement, filtre certaines fréquences de bruits de fond et retransmet les sons filtrés et amplifiés à l’oreille moyenne des personnes malentendantes.
Conclusion
La plupart de ces stratégies de thérapie génique sont actuellement au stade clinique ou en passe d’y arriver 2. Certaines sont d’ores et déjà validées cliniquement et commercialisées, comme le Luxturna qui possède une autorisation de mise sur le marché européen.
Les améliorations des thérapies géniques, tant au niveau de la précision, que de l’efficacité ou de la sécurité offrent de grands espoirs dans le traitement des maladies dégénératives de la rétine au sens large et pas uniquement pour les rétinopathies d’origine génétique. En effet, les stratégies d’expression de facteurs trophiques, ou encore l’optogénétique proposent des mécanismes suffisamment ubiquitaires pour être envisagés comme traitement dans les dégénérescences rétiniennes liées à l’âge et l’environnement, comme la DMLA.