

La paléoprotéomique, une discipline émergente qui analyse les protéines anciennes, offre de nouvelles perspectives fascinantes en paléoanthropologie, l’étude des hominidés éteints. En 2023, une équipe internationale a réussi à séquencer des peptides vieux de 2 Ma, extraits de dents de Paranthropus robustus. Ces séquences révèlent des informations sur la diversité génétique et le sexe des individus étudiés, ainsi que sur la position phylogénétique de cette espèce dans la lignée humaine.
La compréhension de l’évolution humaine est un domaine riche qui combine diverses disciplines scientifiques. Les techniques moléculaires viennent aujourd’hui compléter les méthodes plus traditionnelles d’analyse morphologique des fossiles. Fin 2013, le séquençage presque complet du génome de l’homme de Néandertal 1 a mis en lumière de manière spectaculaire la toute jeune discipline de la paléogénomique en montrant qu’elle pouvait apporter de précieuses informations sur notre histoire ancienne, par exemple en apportant des preuves d’hybridations entre humains archaïques et modernes, ou encore en permettant la découverte d’une toute nouvelle espèce, l’homme de Denisova, à partir de seulement quelques dents et d’un petit bout de phalange 2. Cet exploit technique a ainsi valu à Svante Pääbo de recevoir en 2022 le prix Nobel de physiologie ou médecine « pour ses découvertes sur les génomes d’espèces humaines éteintes et l’évolution humaine » 3.
Contrairement à l’ADN, qui se dégrade rapidement (Figure 1), les protéines se révèlent plus résistantes et sont ainsi utilisées en paléoprotéomique, une discipline qui a pour objectif de faire parler les protéines anciennes, en particulier quand l’ADN ancien a disparu ou est devenu complètement inexploitable. C’est par exemple par l’analyse des protéines anciennes que la mandibule de Xiahe – trouvée dans la grotte karstique de Baishiya dans le district de Xiahe, dans la province de Gansu, en Chine – a pu être attribuée à l’Homme de Denisova, alors qu’aucune trace d’ADN ancien n’avait été préservée 1. La mandibule de Xiahe a été datée d’environ 160 000 ans, mais le champ d’étude de la paléoprotéomique s’est dernièrement étendu à des restes fossiles bien plus anciens, tels que ceux du genre Paranthropus 2.
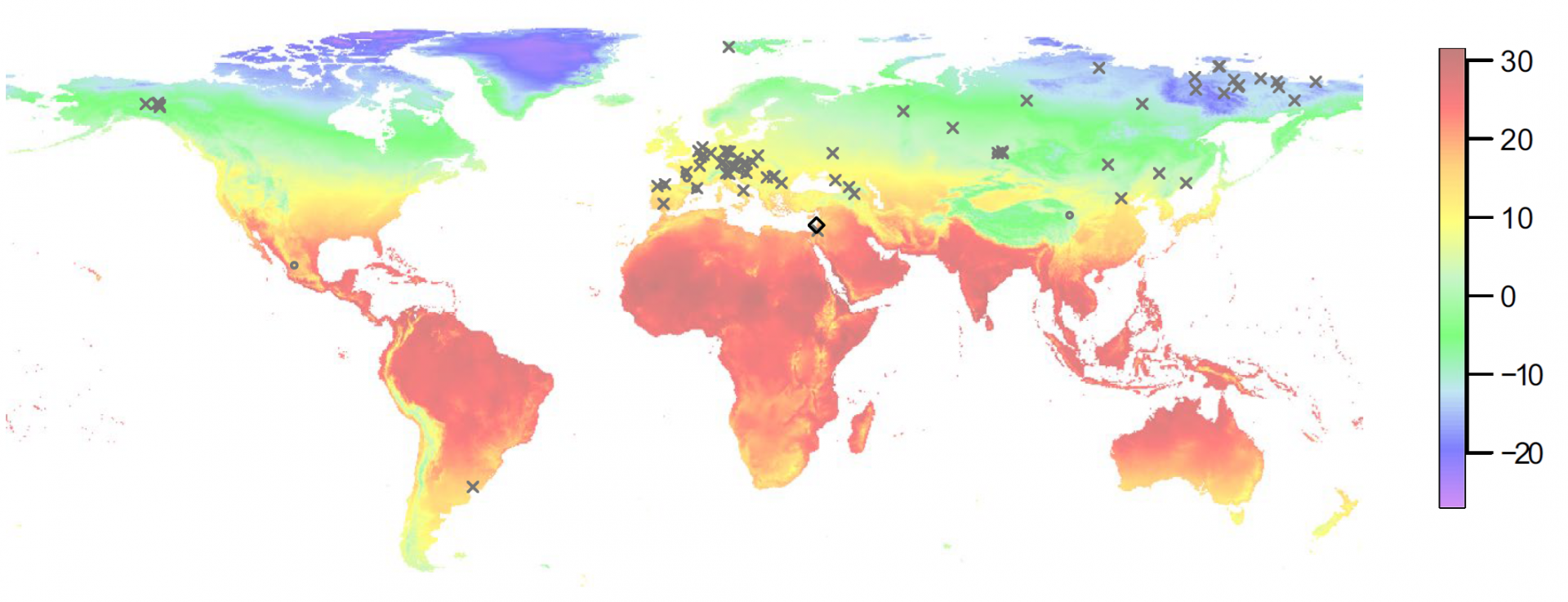
Les croix marquent les sites où l’ADN a été extrait de squelettes, les ronds marquent les sites où l’ADN a été extrait de sédiments. Le losange noir marque la grotte de Sefunim. Les fragments d'ADN les plus anciens jamais séquencés viennent du Groenland et datent de 2 Ma. En dehors du permafrost il n’est pas possible de récupérer de l’ADN plus ancien que 400 ka environ. Les couleurs de la carte montrent les températures moyennes (°C) telles que mesurées entre les années 1970 et 2000.
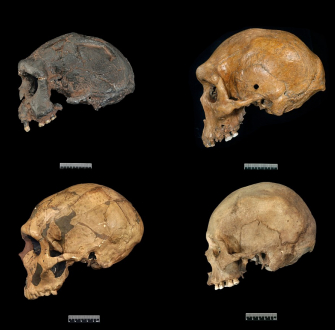
Les paranthropes forment un groupe d’hominidés éteints ayant vécu en Afrique il y a 1 à 2,8 millions d’années. Trois espèces sont généralement distinguées : P. robustus, P. boisei et P. aethiopicus (mais il est possible que ces deux dernières soient en réalité la même espèce). Les paranthropes sont connus pour leur morphologie robuste, avec notamment une crête sagittale proéminente, des mâchoires puissantes et des dents très larges, avec un émail très épais, adaptées à un régime alimentaire herbivore abrasif (Figure 2). Les relations phylogénétiques entre les paranthropes et les autres hominidés, tels que les australopithèques et les premiers membres du genre Homo, sont encore sujettes à débat. Le genre Paranthropus, avec ses caractéristiques morphologiques uniques et sa position controversée au sein de la lignée humaine, offre un terrain idéal pour illustrer l’apport des nouvelles technologies à la recherche paléoanthropologique. C’est dans ce contexte que s’inscrivent les travaux menés par Palesa P. Madupe, Claire Koenig, Ioannis Patramanis et leurs collègues sur une population ancienne de Paranthropus robustus 1.
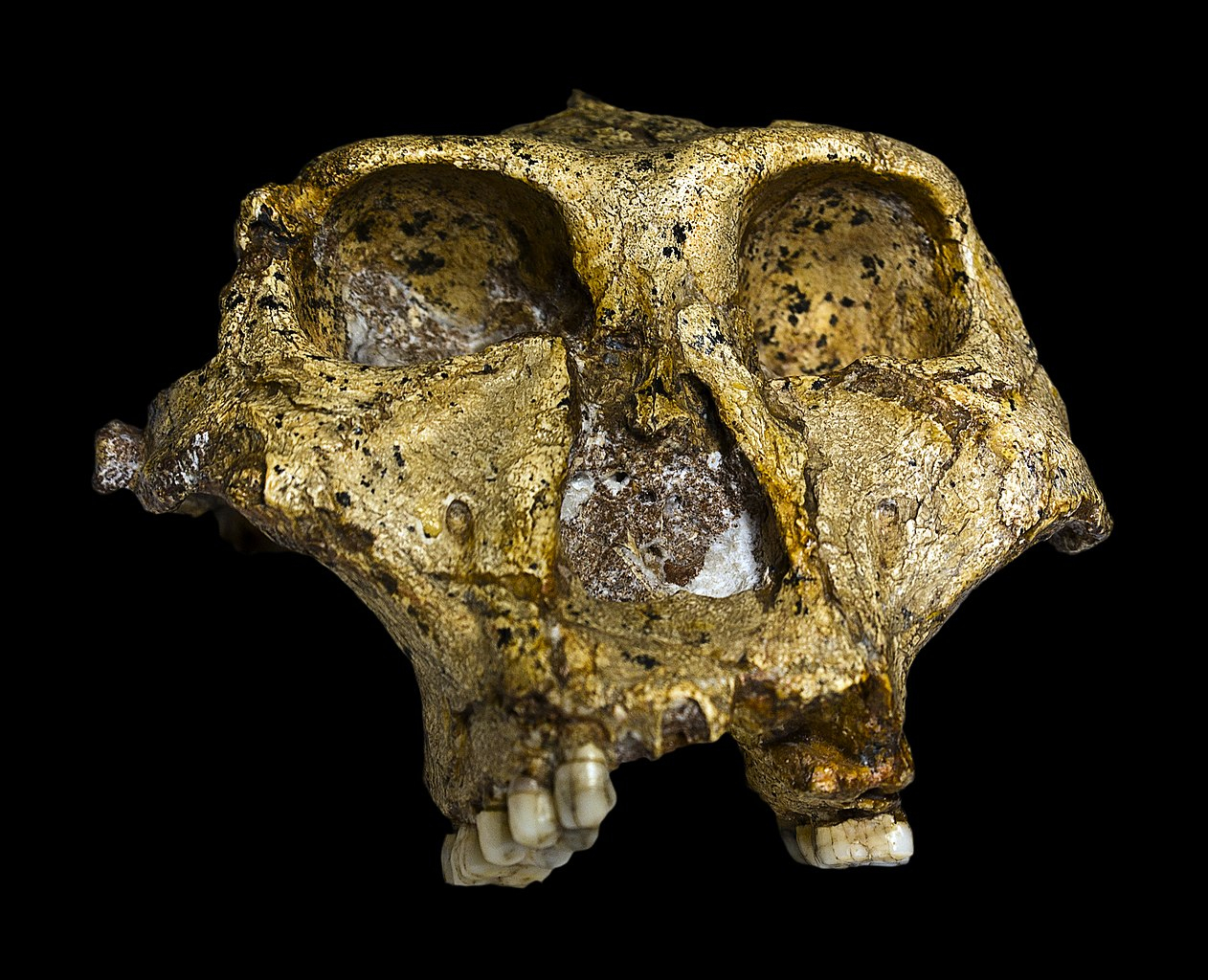
Le crâne original complet (sans la mandibule) de 1,8 million d'années de Paranthropus robustus (SK-48 provenant de Swartkrans (26°00'S 27°45'E), Gauteng), découvert en Afrique du Sud. Collection du Transvaal Museum, Northern Flagship Institute, Pretoria, Afrique du Sud.
Matériels et méthodes
L’objectif principal de l’étude de Madupe et collaborateurs est de clarifier les relations évolutives entre Paranthropus robustus et les autres hominidés éteints, en complétant les analyses morphologiques traditionnelles par des données moléculaires. Pour ce faire les chercheurs ont analysé quatre spécimens dentaires (Figure 3) attribués à P. robustus trouvés sur le site de Swartkrans et datant d’environ 2 millions d’années (étage du Gélasien, Pléistocène inférieur).
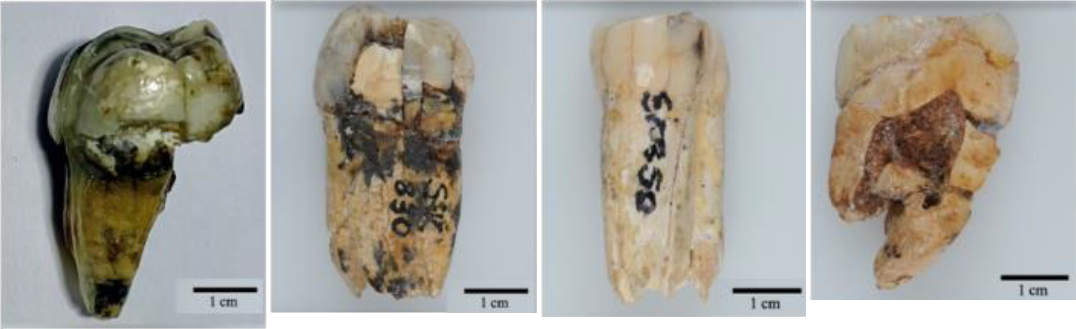
Les spécimens ont été trouvés dans la grotte de Swartkrans en Afrique du Sud à 40 km au Nord-Ouest de Johannesbourg. Les dépôts sédimentaires contenant ces fossiles sont datés d’environ 1,8 à 2,2 Ma. Ces spécimens étaient conservés au Musée national Ditsong d’Histoire naturelle. De gauche à droite : SK 835, une 3e molaire supérieure gauche ; SK 830 une 4e prémolaire inférieure gauche ; SK 850, une 3e prémolaire inférieure droite ; SK 14132, une 3e molaire supérieure droite. Notez que les deux prémolaires humaines sont numérotées 3 et 4 parce que les prémolaires 1 et 2 ont été perdues au cours de l’évolution.
L’émail dentaire a été réduit en poudre et dissout avec de l’acide trifluoroacétique pour en extraire les peptides anciens. Cette opération a été réalisée à l’université de Copenhague dans un laboratoire spécialement équipé pour réduire les risques de contamination par des peptides modernes. Après nettoyage, les peptides ont été séparés par chromatographie en phase liquide, puis analysés par spectrométrie de masse en tandem (LC-MS/MS). Cette technique consiste dans un premier temps à ioniser les peptides, puis à mesurer leurs rapports masse/charge. Dans un deuxième temps les peptides sont fragmentés par des collisions avec un gaz inerte et les rapports masse/charge de ces fragments sont à leur tour mesurés (d’où le terme « en tandem »). Le traitement des données ainsi récoltées permet de déterminer la séquence du peptide analysé.
Ces séquences peptidiques ont ensuite été minutieusement comparées et alignées avec les protéines connues de l’émail dentaire chez différentes espèces de singes plus ou moins éloignées de l’homme moderne. Des arbres phylogénétiques ont été produits pour chaque protéine individuellement et pour une concaténation de toutes les protéines (c’est-à-dire toutes les séquences alignées mises bout à bout). Les chercheurs n’ont pas utilisé le critère traditionnel du maximum de parcimonie pour construire ces arbres mais deux méthodes probabilistes plus sophistiquées : le maximum de vraisemblance et l’inférence bayésienne (voir l'encadré ci-dessous). L’étude inclut également une analyse morphométrique des dents de Swartkrans ainsi que de dents provenant d’autres sites.
Les critères d’optimalité d’un arbre phylogénétique
Il existe différents critères permettant d’évaluer la justesse de plusieurs arbres phylogénétiques en concurrence et ainsi de choisir le meilleur. Dans les cas les plus complexes, il est nécessaire de recourir aux critères les plus sophistiqués.
1. Maximum de parcimonie (MP)
Le principe de parcimonie énonce que le meilleur arbre est celui qui explique les données (les séquences observées) avec le plus petit nombre total de changements évolutifs (les mutations). Le scénario le plus parcimonieux est privilégié, ce qui revient à chercher l’arbre le plus court si les longueurs des branches sont proportionnelles au nombre de transformations qu’elles accueillent.
Avantages : Assez rapide, facile à comprendre.
Limites : Non fiable. Il est malheureusement démontré théoriquement et empiriquement que ce critère n’est pas statistiquement cohérent 123. Cela signifie que les méthodes utilisant ce critère convergent vers un arbre incorrect même si la quantité de données disponibles tend vers l’infini. Cela est dû à différents biais statistiques, liés notamment au fait qu’une même position peut muter plusieurs fois au cours de l’évolution. La quantité d’homoplasies est ainsi systématiquement sous-estimée.
2. Minimum d’évolution (ME)
Le principe d’évolution minimale ressemble beaucoup au critère précédent : le meilleur arbre est le plus court 4. Mais les longueurs des branches ne sont pas ici estimées par le nombre minimum de mutations accommodant la topologie de l’arbre. Les longueurs des branches sont calculées à partir des distances génétiques entre toutes les paires de séquences, et ces distances génétiques sont elles-mêmes le résultat d’une estimation statistique du nombre de mutations qui se sont réellement produites le long du chemin reliant deux séquences, et non simplement le nombre de mutations observées. Ces estimations des distances génétiques reposent sur des modèles stochastiques d’évolution des séquences. Le Neighbor-Joining est l’algorithme heuristique le plus connu implémentant ce principe d’évolution minimale, ou plus exactement le critère BME (Balanced Minimum Evolution). Le critère BME correspond à l’ajout d’une pondération des distances génétiques lors des calculs des longueurs des branches pour une topologie particulière, ce qui augmente les performances 5.
Avantages : Efficace et très rapide pour traiter de grandes quantités de données.
Limites : Bien que fiable (statistiquement cohérent) et robuste (résistant à d’importantes perturbations des données), ce critère n’est pas aussi puissant que des méthodes statistiques plus sophistiquées.
3. Maximum de vraisemblance (ML)
La méthode du maximum de vraisemblance cherche l’arbre qui maximise la probabilité d’observer les données (les séquences) avec un certain modèle d’évolution 678. L’adéquation du modèle aux données est elle-même évaluée par cette méthode d’inférence phylogénétique. Le maximum de vraisemblance est un critère bien connu des statisticiens, mais son interprétation est peu intuitive. Il s’agit en effet d’un calcul de la probabilité des données, et non de la probabilité de l’arbre. Cela ne garantit pas que l’arbre choisi est le plus probable. En particulier, la somme des vraisemblances de tous les arbres possibles n’est pas égale à 1. Autrement dit, cette méthode repose sur le paradigme fréquentiste, et cela pose les mêmes problèmes d’interprétation qu’une valeur p, ou qu’un intervalle de confiance par exemple. Calculer l’intervalle de confiance à 95 % d’une variable x ne signifie pas que x a 95 % de chances d’être dans cet intervalle. Cela signifie que, si l’on répétait l’expérience de nombreuses fois et calculait un nouvel intervalle de confiance à chaque fois, 95 % de ces intervalles contiendraient la valeur réelle de x.
Avantages : Calculs probabilistes explicites et réalistes prenant en compte toutes les informations disponibles.
Limites : Plus complexe et coûteux en temps de calcul, paradigme fréquentiste rendant l’interprétation difficile.
4. Maximum à postériori (MAP) / Inférence bayésienne
Dans le paradigme bayésien on cherche l’arbre ayant la probabilité la plus élevée. Cette probabilité est dite à postériori, car elle est calculée en tenant compte à la fois des données observées et d’une distribution à priori des probabilités. La somme des probabilités de tous les arbres possibles est bien égale à 1. De plus, cette méthode intègre l’incertitude sur les paramètres du modèle en les marginalisant, c’est-à-dire en considérant toutes les valeurs possibles de ces paramètres pondérées par leur probabilité : taux de mutation, longueurs de branches, etc. C’est un avantage considérable par rapport au maximum de vraisemblance puisqu’avec cette dernière on ne calcule pas la probabilité des données étant donné la topologie de l’arbre, mais la probabilité des données étant donné la topologie et les valeurs optimisées des différents paramètres. C’est ce qu’on appelle une probabilité conjointe. Or la détermination de ces paramètres n’est pas l’objectif recherché. Le but poursuivi est d’utiliser ces paramètres pour déterminer la topologie de l’arbre et c’est ce tour de force que parvient à effectuer l’inférence bayésienne : extraire la probabilité d’une topologie seule pour toutes les distributions estimées de tous les paramètres. Mais cette prouesse n’est possible que grâce à de très lourdes simulations, il est absolument impossible de calculer ces probabilités de manière analytique 9.
Avantages : Évaluation explicite de la probabilité marginale d’un arbre rendant l’interprétation simple, incorpore toutes les données disponibles dans un modèle réaliste.
Limites : Exige des temps de calcul considérables reposant sur des simulations.
Résultats et discussion
L’extraction des protéines de paranthropes et la détermination de leurs séquences a été un succès. Le transport des restes au cours de crues subites et le dépôt des sédiments au sein d’une grotte dans une zone au climat aride a certainement contribué à leur bonne préservation en limitant les oscillations thermiques. Les protéines ainsi récupérées sont principalement des protéines structurales de la matrice extracellulaire de l’émail dentaire telles que l’amélogénine, l’améloblastine, l’énaméline, le collagène, etc. L’analyse détaillée des séquences montre des différences génétiques entre les individus et même de l’hétérozygotie (deux allèles différents chez un même individu).
Il a également été possible d’identifier le sexe des quatre spécimens. En effet, l’amélogénine possède deux isoformes, l’une produite par le gène AMELX porté par le chromosome X, l’autre produite par le gène AMELY porté par le chromosome Y. L’amélogénine‑X et l’amélogénine‑Y n’ont pas tout à fait la même séquence et peuvent être distinguées. Comparez par exemple ce peptide dans l’amélogénine‑X « YQSIRPPYPS » avec celui correspondant dans l’amélogénine‑Y « YQSMIRPPYSS ». L’identification de ces peptides spécifiques de l’amélogénine‑Y chez deux spécimens a permis de les identifier comme des mâles (SK 850 et SK 835) tandis que les deux autres sont très probablement des femelles (SK 830 et SK 14132). L’individu SK 835 avait précédemment été identifié comme une femelle sur la base de sa petite taille, ce qui met en lumière les limites des méthodes de sexage fondées sur le dimorphisme sexuel. Il faut toutefois noter que le gène AMELY est parfois absent (délétion complète) chez certains mâles comme cela a été rapporté pour Homo sapiens et pour Homo neanderthalensis. Cette technique moléculaire pourrait donc faussement identifier des mâles comme étant des femelles dans de rares cas.
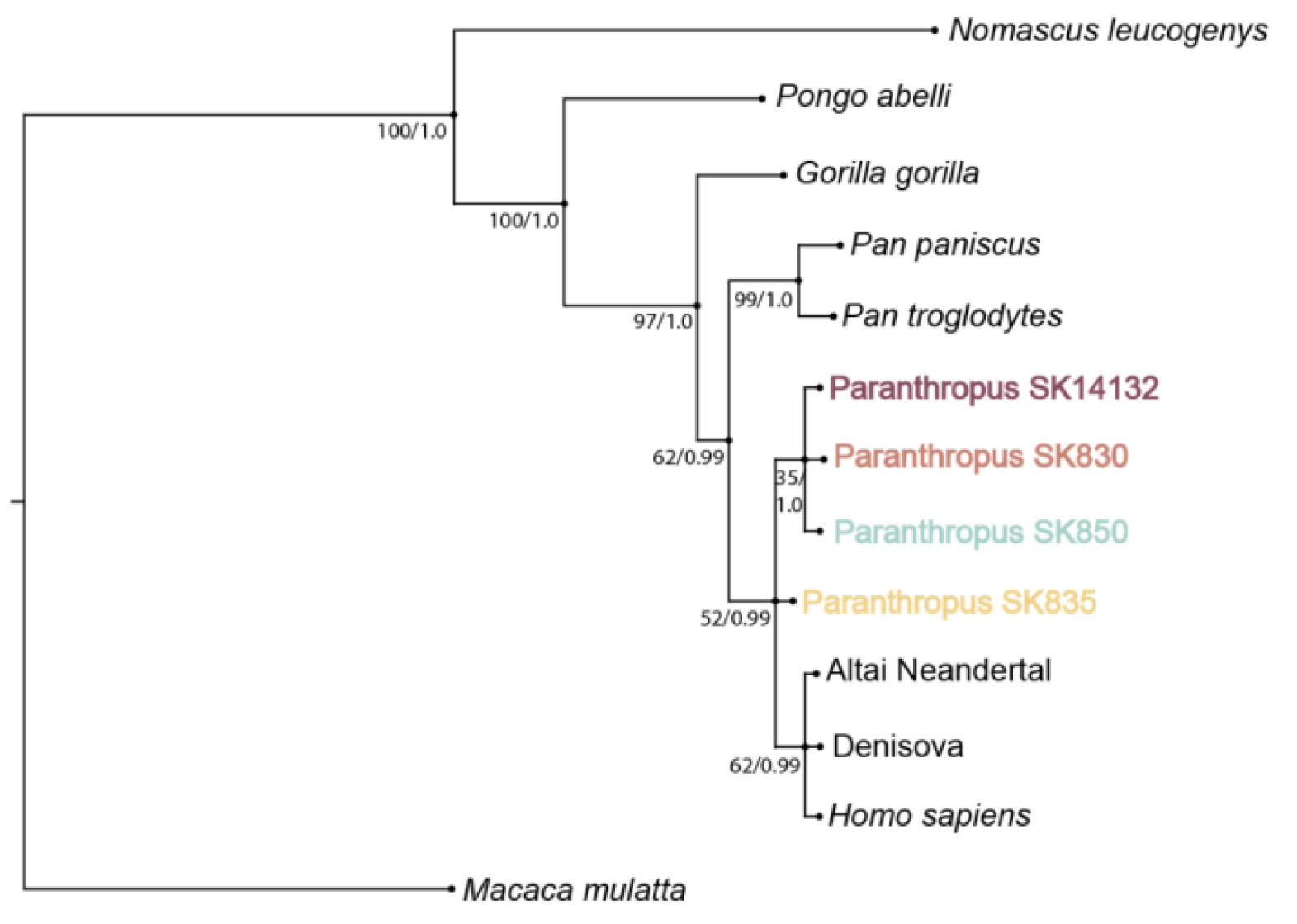
Les nombres aux nœuds représentent à gauche le bootstrap de 0 à 100 des réplicats de l’analyse par maximum de vraisemblance et à droite la probabilité à postériori de 0 à 1 de l’analyse par inférence bayésienne. Le bootstrap est une mesure permettant d’évaluer la stabilité d’un clade lorsque l’analyse est effectuée à plusieurs reprises (généralement au moins 1000 fois). Pour chaque réplicat on rééchantillonne aléatoirement les données, c’est-à-dire qu’on tire au sort (avec remise) les positions des résidus dans l’alignement des séquences qui sont conservées ou éliminées. Cela revient à perturber les données en attribuant un poids aléatoire à ces différentes positions, puisque certaines peuvent ne pas être choisies alors que d’autres peuvent être choisies une ou plusieurs fois. On observe alors le pourcentage de réplicats dans lesquels le meilleur arbre contient le clade d’intérêt. Un score de bootstrap faible (inférieur à 80 %) est le signe d’une grande sensibilité des données aux perturbations. Cela ne signifie pas que ces clades sont nécessairement faux, ni même improbables, mais cela rend leur position moins fiable, on ne peut pas leur accorder la même confiance qu’à ceux qui sont plus stables.
La reconstruction phylogénétique (Figure 4) montre que tous les spécimens de P. robustus sont plus proches du genre Homo que des autres grands singes, confirmant ainsi l’appartenance des paranthropes à la lignée humaine. Ce résultat est bien sûr cohérent avec les meilleures phylogénies de la lignée humaine disponibles actuellement, comme celle présentée dans la Figure 5 12.
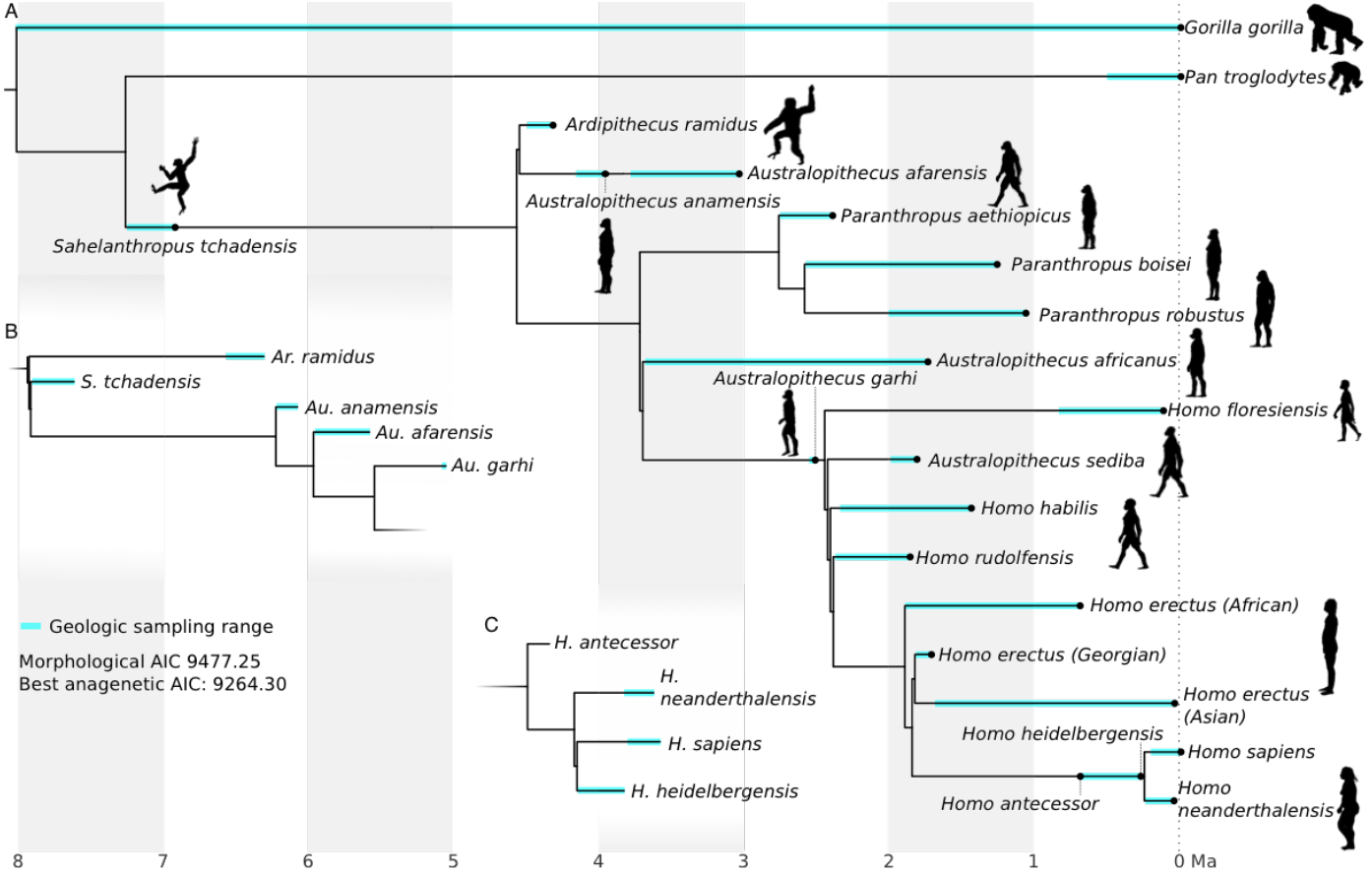
Cet arbre représente la meilleure estimation de Parins-Fukuchi et coll., 2018. À la différence d’autres méthodes de construction d’arbres phylogénétiques, n’utilisant que des données morphologiques, les auteurs estiment, dans l’étude dont est tirée cette figure, qu’il est préférable de recourir en plus aux données de datations des fossiles (lignes bleues) pour proposer des relations d’ancêtres à descendants. C’est ainsi qu’ils identifient Sahelanthropus tchadensis comme l’ancêtre des Hominines ultérieurs, Australopithecus anamensis comme l’ancêtre d’Australopithecus afarensis, Australopithecus garhi comme l’ancêtre du genre Homo, Homo antecessor comme l’ancêtre d’Homo heidelbergensis, qui aurait lui même donné Homo sapiens et Homo neanderthalensis. Les sous-figures B et C montrent des portions de l'arbre reconstruites sans proposer de relations d’ancêtres à descendants et qui, selon les auteurs, induisent des « résultats erronés ». Les silhouettes viennent du site PhyloPic.
Trois des quatre spécimens de paranthropes forment un clade, mais la position de SK 835 est plus incertaine car la trichotomie que l’on observe indique que ce nœud n’a pas pu être résolu. Autrement dit l’algorithme employé pour construire cet arbre a évalué qu’aucune des trois dichotomies possibles n’était significativement plus vraisemblable que les deux autres.
Différents scénarios, cohérents avec chacune de ces trois possibilités, peuvent être imaginés. Cependant, avant de surinterpréter ces résultats, il faut se rappeler qu’ils reposent sur assez peu de données. L’ensemble des peptides récoltés chez les paranthropes ne comprend en effet que 17 positions cladistiquement informatives à l’échelle des grands singes, et seulement 2 positions qui sont différentes de ce que l’on rencontre dans le genre Homo. La première différence se situe en position 637 dans l’alignement de la protéine COL17A1 pour SK 835 et SK 14132. Ces deux spécimens ont l’état ancestral (proline) qu’ils partagent avec tous les autres grands singes en dehors de la lignée humaine, tandis que le genre Homo a l’état dérivé (alanine). Le peptide en question n’a pas pu être détecté chez les deux autres spécimens. La deuxième différence se situe en position 137 dans l’alignement de la protéine ENAM. Les grands singes africains et le genre Homo ont ici une arginine tandis que SK 830 et SK 850 ont une glutamine, SK 835 une arginine, et SK 14132 est hétérozygote avec les deux versions du peptide. Les orangs-outans présentent aussi une glutamine à la place de l’arginine, ce qui indique une homoplasie, ici une convergence évolutive. Il faut donc se garder de toute interprétation hâtive.
Le polymorphisme génétique est une autre difficulté importante qu’il faut souligner dans ce genre d’études. Le spécimen SK 14132 démontre que plusieurs allèles différents d’un même gène peuvent exister dans une population ancienne, comme c’est le cas dans les espèces modernes. La question du choix de l’allèle pour effectuer la reconstruction phylogénétique se pose alors naturellement. Idéalement, les séquences comparées entre deux espèces doivent avoir divergé après la séparation des deux lignées. On dit dans ce cas-ci que les séquences sont orthologues. Si les séquences comparées ont commencé à diverger avant la séparation des lignées cela signifie qu’elles descendent d’allèles différents présents chez le dernier ancêtre commun. On dit dans ce cas-là que les séquences sont paralogues. Un arbre de gènes ne sera identique à l’arbre des espèces que si l’on compare des allèles orthologues, c’est-à-dire issus d’un même allèle ancestral dont la séquence a divergé entre les espèces après chaque spéciation. Si on compare des allèles paralogues, c’est-à-dire des séquences ayant divergé avant les spéciations qu’on cherche à étudier, alors l’arbre des espèces sera biaisé (Figure 6).
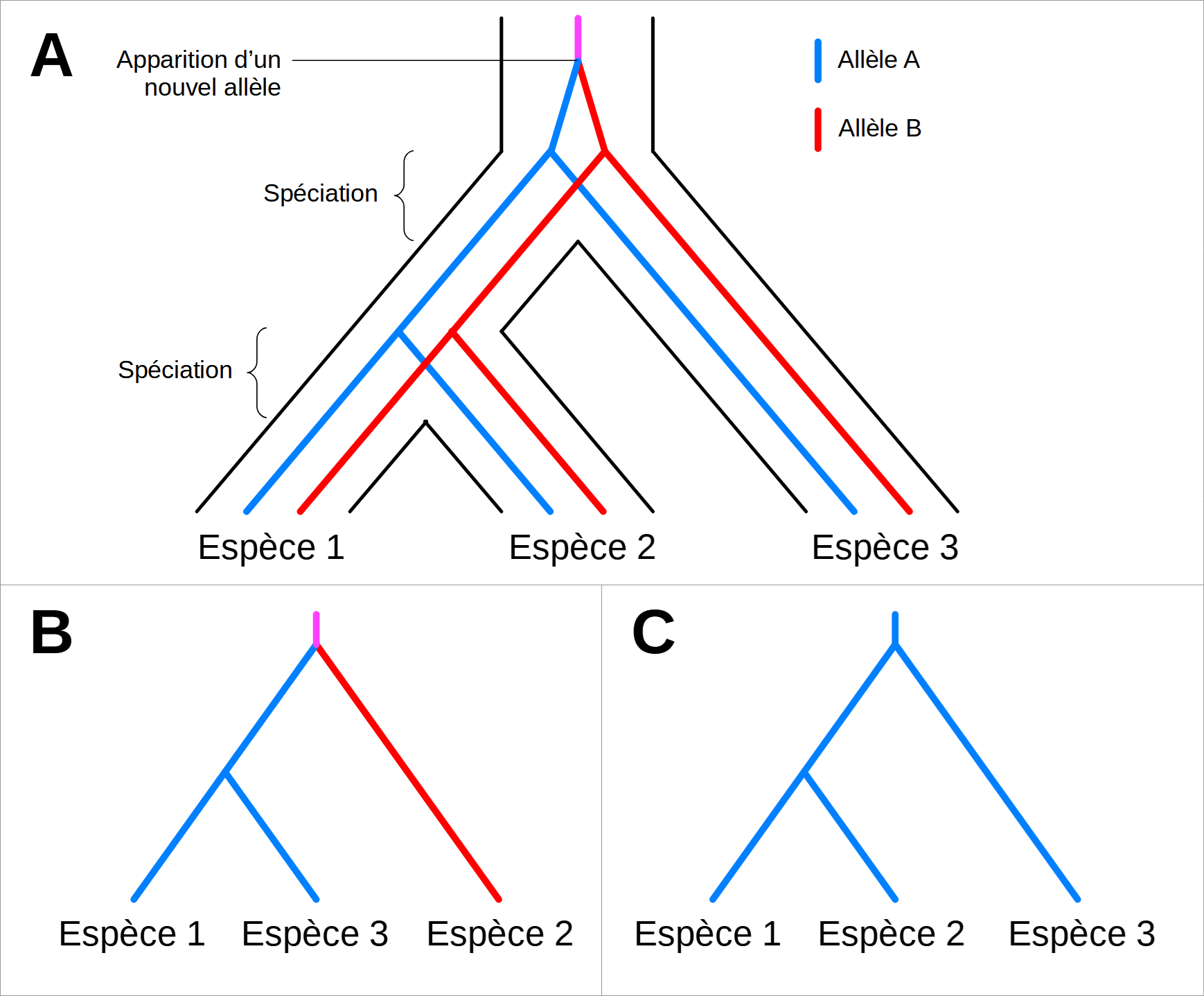
A. Histoire phylogénétique réelle d’une lignée montrant les évènements de spéciation et de diversification génétique avec l’exemple des allèles A et B issus d’un même allèle ancestral. Les allèles A et B sont deux versions d’un même gène qui coexistent chez les trois espèces.
B. Arbre phylogénétique des espèces biaisé, construit avec les séquences des allèles A des espèces 1 et 3, et la séquence de l’allèle B de l’espèce 2. Les allèles A et B sont paralogues.
C. Arbre phylogénétique des espèces non biaisé, construit avec les séquences des allèles A des trois espèces. Les différentes versions de l’allèle A sont orthologues.
Cette paralogie n’est pas facile à identifier, même avec un échantillonnage plus large des populations, car le polymorphisme ancestral peut disparaître chez les descendants. L’un ou l’autre allèle peut alors s’être fixé aléatoirement dans les espèces modernes. Ce phénomène porte le nom de tri incomplet des lignées, ou incomplete lineage sorting (ILS). L’inférence d’un arbre à partir de séquences paralogues reflète alors l’histoire de ces séquences, mais pas nécessairement l’histoire des espèces. L’apparition d’une discordance due à ce biais est d’autant plus probable que les espèces sont apparentées et que les évènements de spéciations se sont succédé rapidement.
Ces distorsions peuvent être rectifiées en échantillonnant plusieurs gènes différents comme l’ont fait Madupe et ses collaborateurs. Dans leur étude, on remarque justement que les arbres phylogénétiques produits à partir des protéines individuelles sont tous différents les uns des autres et sont également différents de celui produit à l’aide de toutes les séquences concaténées. La concaténation des séquences de plusieurs gènes ou protéines permet ainsi de compenser, au moins partiellement, ce type de biais. Notons pour finir qu’il existe des méthodes plus complexes, mais statistiquement plus robustes que la simple concaténation, fondées sur la théorie de la coalescence, pour tenir compte de ce phénomène, mais elles n’ont pas été employées dans les travaux présentés ici.
Conclusion
On peut regretter que ces travaux ne soient pas parvenus à récolter davantage de données moléculaires, comme des peptides plus longs comportant davantage de positions cladistiquement informatives, ou un échantillonnage d’une plus grande diversité de spécimens, comme d’autres espèces de paranthropes ou des australopithèques. Les méthodes employées dans cette étude constituent toutefois une véritable percée qui ouvre de nouvelles perspectives pour la recherche en paléoanthropologie. Il est très probable que les mêmes équipes, et d’autres, soient déjà en train de réaliser de nouvelles investigations afin d’approfondir cette voie prometteuse.
Une étude a d’ailleurs permis d’évaluer explicitement le potentiel des protéines anciennes pour l’inférence phylogénétique 1. Les chercheurs ont utilisé les séquences connues d’une grande variété de primates actuels et ont simulé les dégradations typiques qu’elles subissent. Ils ont ensuite vérifié que l’arbre phylogénétique reconstruit à partir de ces séquences très parcellaires avait bien une topologie similaire à l’arbre de référence. Les résultats montrent que la plupart des familles et des genres sont placés correctement avec des âges simulés d’environ 1-2 Ma pour toutes les séquences. De plus, une espèce peut être placée correctement dans l’arbre avec un âge simulé de 5 Ma de ses séquences si les séquences des autres espèces sont complètes (ce qui est typiquement ce qu’on cherche à faire quand on veut placer un fossile au sein d’une phylogénie connue d’espèces actuelles). Ces résultats sont donc encourageants. Par conséquent on peut s’attendre à de nouvelles découvertes dans les années à venir grâce à ces méthodes novatrices.


