Après avoir brièvement rappelé les mécanismes génétiques à l'origine des cancers, ainsi que le phénomène de l'apoptose, cet article présente deux gènes suppresseurs de cancer, BIM et p53, et précise quelles sont les voies de recherche en cours ayant pour objectif d'amplifier leurs effets sur les cellules.
Introduction
Les cancers sont caractérisés par une multiplication anarchique de cellules [1]. Ils naissent de mutations variées, qui s’accumulent et se combinent au fil du temps et qui touchent :
-
Les proto-oncogènes ; ces mutations transformant ces gènes aux fonctions parfaitement physiologiques en leurs versions oncogènes, responsables d’une stimulation incontrôlée des divisions cellulaires. Plus de 100 proto-oncogènes étaient déjà identifiés en février 2005 [2].
-
Les gènes suppresseurs de tumeur actifs ; ces mutations transforment ces gènes aux fonctions également parfaitement physiologiques, en leur versions inactives, incapables d’inhiber les divisions cellulaires. En novembre 2003, 174 de ces gènes figuraient déjà sur une base de données.
La liste des proto-oncogènes et des gènes suppresseurs de tumeur ne cesse de s’allonger au fur et à mesure que de nouvelles fonctions de gènes sont découvertes.
Redonner aux cellules cancéreuses la capacité de contrôler leurs divisions serait un bon moyen de lutter contre leur prolifération. C’est ce qui a été tenté chez la souris au tournant de l’année 2007, avec les gènes suppresseurs de tumeur Bim et p53. Que nous enseignent ces expériences ?
Bim, un gène suppresseur de tumeur
L'apoptose ou mort cellulaire programmée
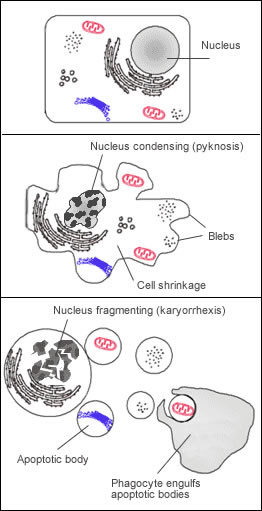
L’apoptose est un mécanisme génétiquement contrôlé, aboutissant à la destruction d’une cellule via, entre autres : la condensation de la chromatine et la fragmentation de l’ADN, le désassemblage de la membrane nucléaire, la condensation du cytoplasme et sa fragmentation en corps apoptotiques (délimités par la membrane plasmique) et enfin l’élimination de ces corps apoptotiques par phagocytose, réalisée par les cellules voisines ou des macrophages (voir figure ci-contre). Le tout n’entraîne aucune inflammation ni aucune perturbation des cellules environnantes, au contraire de la mort cellulaire par nécrose ou par sénescence.
L’apoptose peut être suivie non seulement sur ces critères morphologiques, mais également sur des critères biochimiques :
-
Activation de caspases (Cystein ASPartic acid proteASE ou protéase contenant une cystéine et clivant après un aspartate) ; 14 caspases découvertes à ce jour dans l’espèce humaine), enzymes qui agissent en réseau et s’activent en cascade. La caspase-3, commune à de nombreux sentiers d’apoptose, joue un rôle pivot. Signalons qu’il existe des apoptoses qui n’utilisent pas ces caspases.
-
Clivage de protéines cibles spécifiques par les caspases, donc après un acide aspartique, comme les lamines qui composent le cytosquelette nucléaire.
-
Découpage de l’ADN en petits fragments de 180 paires de bases, en moyenne. Ces fragments sont obtenus par action d’une DNAse agissant prioritairement au niveau des zones de plus grande accessibilité de l’ADN, à savoir les parties situées entre les nucléosomes.
Bim et ses propriétés
Le gène Bim est un gène pro-apoptotique. Il appartient à la famille des gènes Bcl-2, qui comporte aussi bien des membres aux propriétés anti-apoptotiques (donc des oncogènes) que des membres aux propriétés pro-apoptotiques (donc des suppresseurs de tumeur) et qui interagissent entre eux.
S’il reste encore des zones d’ombre concernant les modes d’action de Bim (qui emprunterait plusieurs voies), il semble établi que la protéine Bim peut se lier, via un domaine nommé H3, aux membres anti-apoptotiques de la famille des protéines Bcl-2, et ainsi inhiber leur action.
Fournir le peptide Bim
Bim est muté dans de nombreux cancers, et les cellules, qui ne sont plus éliminées, prolifèrent. Leur fournir la version non mutée de Bim, sous forme du gène ou de sa protéine, pourrait permettre de lutter contre la perte de contrôle de leur activité mitotique.
L’équipe pluridisciplinaire de William Hawkins aux États-Unis a eu l’idée d’utiliser certaines propriétés du virus du sida pour construire un véhicule approprié : le peptide chimère TAT-Bim [3, 4]. L’intérêt du peptide TAT, dérivé de la protéine TAT virale, est qu’il est capable de perforer les nombreuses cellules cibles du virus et de tracter à l’intérieur de celles-ci ce qui lui est lié, en l’occurrence ici, le domaine H3 de la protéine Bim non muté, aux propriétés convoitées.
Le travail a été mené in vitro sur des lignées de cellules tumorales dérivées de lymphome, de mélanome et de cancer pancréatique ; et in vivo sur des souris rendues cancéreuses par implantation sous cutanée de deux de ces lignées, issues de cancer pancréatique ou de mélanome.
In vitro , le travail a fourni différentes informations sur ce peptide TAT-Bim :
-
Il pénètre bien dans les trois lignées tumorales testées, comme en témoigne un double marquage de TAT et de Bim.
-
Il exerce une action apoptotique, comme en atteste la mise en évidence d’une activité caspase 3.
-
Il exerce cette action apoptotique via Bim, puisque l’utilisation d’un peptide chimère TAT-Bim comprenant un Bim inactif n’a aucun effet délétère sur les cellules, pas plus que TAT seul.
-
Il augmente en outre l’efficacité des radiothérapies, dont l’action pro-apoptotique s’exerce d’autant mieux que l’un des sentiers apoptotiques a été reconstitué.
In vivo, le travail effectué sur les souris porteuses des redoutables tumeurs pancréatiques ou issues de mélanome a connu un début de succès :
-
Après 40 jours d’injection de TAT-Bim dans les tumeurs, 80 % des souris survivent, au lieu de 20 % dans les lots non traités.
-
Cette protection exige la présence continue de TAT-Bim, puisque trois à cinq jours après l’arrêt du traitement les tumeurs recommencent à grossir.
-
Enfin, comme avec les cultures cellulaires, l’efficacité des radiothérapies augmente lorsque les souris sont traitées par TAT-Bim.
Les chercheurs prévoient de lier à TAT-Bim des molécules reconnaissant spécifiquement les cellules cancéreuses, pour épargner les cellules saines. Et comme le petit peptide TAT est capable de tracter des molécules mille fois plus grosses que lui, ils projettent aussi de lui lier bien d’autres molécules curatives.
Le gène p53, un autre gène suppresseur de tumeur
Le gène p53 et ses propriétés
Ce gène p53 remplit des rôles multiples dans la cellule, via le facteur de transcription dont il gouverne la synthèse : la protéine p53 [5].
-
Rôle dans l’homéostasie cellulaire, comme tout gène suppresseur de tumeur, en contrebalançant les ordres de division délivrés par les proto-oncogènes.
-
Rôle de gardien du génome, par blocage transitoire du cycle cellulaire en cas de mutation, donnant ainsi le temps nécessaire à une réparation ; ou, lorsque celle-ci est impossible, par blocage permanent du cycle et donc des divisions qui pourraient propager ces mutations ; ou encore, par mise à mort des cellules mutées, au travers de l’apoptose, ou d’autres types de mort cellulaire [6]. Si dans une cellule normale, la protéine est peu abondante (demi-vie réduite), sa quantité augmente dans certaines circonstances : cassure de l’ADN, stress, mutations…
Le rôle important de p53 dans le phénomène de cancérogenèse est attesté par le fait que le gène, ou la protéine, est touché dans la plupart des cancers : mutations dans le gène p53, ou inactivation, par exemple par surexpression d’inhibiteurs spécifiques comme MDM2 qui ubiquitinyle p53, ce qui entraîne sa dégradation (l’ubiquitine est un peptide qui, accroché à une protéine, agit comme un signal entraînant la dégradation de cette protéine par les protéasomes). Il s’agit donc d’une piste potentiellement intéressante dans la recherche de nouvelles stratégies de lutte contre cette maladie.
Réactiver le gène p53
Depuis longtemps, les mutations de p53 étaient connues pour participer au déclenchement de cancers variés, mais personne ne savait si elles étaient également nécessaires à leur persistance. Plusieurs équipes ont su répondre à cette question, au tournant de l’année 2007, en construisant – par des méthodes extrêmement sophistiquées – des souris chez lesquelles l’expression du gène p53 pouvait être désactivée, puis à loisir réactivée, par des moyens variés [7, 8].
Le gène p53 et la réactivation de Lowe
L’équipe de Scott Lowe du Cold Spring Harbor à New York a développé une technique à même d’induire un cancer du foie, partiel et très agressif chez la souris, en inactivant l’expression de p53 par interférence ARN [7, 9]. Lorsque l’expression de p53 est réactivée, non seulement les portions tumorales du foie cessent de grossir, mais elles disparaissent complètement : la persistance des tumeurs requiert bien l’inactivation continue de p53.
Contrairement à ce qui était attendu par les auteurs, la disparition de ces tumeurs ne s’effectue pas par apoptose (pas d’intervention de la caspase-3). En effet, dans les expériences réalisées par l’équipe de Lowe, l’effet primaire de la réexpression de p53 a été l’induction du programme de sénescence cellulaire, qui n’est pas une modalité de mort cellulaire mais un état qui se caractérise en particulier par un arrêt de la prolifération cellulaire. L’expression du programme de sénescence cellulaire a entraîné une forte réaction inflammatoire, et la destruction des cellules tumorales a été réalisée par l’intervention du système immunitaire non spécifique.
L’équipe de Lowe poursuit son travail en continuant à explorer les voies de la disparition des cellules tumorales, qui semble donc impliquer une collaboration entre sénescence cellulaire et système immunitaire non spécifique.
Le gène p53 et la réactivation de Jacks
L’équipe de Tyler Jacks du MIT a su, de son côté, réactiver par voie pharmacologique la protéine p53 mutée, chez des souris porteuses de tumeurs du tissu lymphatique (lymphomes) ou du tissu conjonctif (sarcomes) chez qui ce gène avait d’abord été désactivé [8].
Après réactivation de p53 :
-
Lymphomes et sarcomes régressent, ce qui montre que l’inactivation de p53 est indispensable à leur maintenance.
-
Cette régression s’effectue cette fois-ci selon deux voies différentes :
-
une voie rapide, complète, par apoptose pour les lymphomes ;
-
une voie plus lente, qui passe par un blocage du cycle cellulaire avec présence de figures de sénescence.
-
Fait à souligner, l’activation de p53 conduisant à la mort cellulaire n’altère en rien les cellules non tumorales, ce qui n’était pas forcément attendu, puisque chez ces dernières la protéine p53 est également inactive (liée qu’elle est à la protéine inhibitrice MDM2).
Ces résultats enseignent que p53 est non seulement en relation avec les voies de l’apoptose, mais aussi avec celles de la sénescence. Ils invitent à tenter de traiter, un jour, certains cancers humains par réactivation pharmacologique de leur p53 muté.
Le gène p53 et la réactivation d’Evan
L’équipe de Gerard Evan, de l’université de Californie à San Francisco, a testé, elle aussi, l’effet de la restauration de p53 sur un autre type de lymphome murin [10]. Elle a obtenu un début de succès : les souris survivent, leurs cellules tumorales disparaissent par apoptose – il faut donc bien la continuelle inactivation de p53 pour que les tumeurs persistent – mais cette survie ne dure qu’un temps. D’autres mutations sont sélectionnées qui favorisent l’émergence de cellules à nouveau cancéreuses.
Les chercheurs tentent de mettre au point une combinaison de thérapies à même de contrecarrer la grande adaptabilité des cellules cancéreuses.
Conclusion
Bien que le développement des tumeurs cancéreuses dépende d’une combinaison de gènes mutés (proto-oncogènes et gènes suppresseurs de tumeur), leur régression peut être induite – au moins un temps – par le retour à la normale d’un seul d’entre eux. Ainsi en est-il chez la souris, pour deux gènes suppresseurs de tumeurs :
-
Le peptide Bim non muté, tant qu’il est fourni, fait disparaître, par apoptose, un cancer du pancréas et un mélanome.
-
La protéine p53, nouvellement ré-exprimée fait disparaître, au moins temporairement, différents cancers par des voies variées : apoptose pour des lymphomes, sénescence en association avec le système immunitaire non spécifique pour un cancer du foie, simple sénescence pour un sarcome [11].
Mais les cellules cancéreuses ayant une grande propension à muter et, par là, à circonvenir les moyens de défense nouvellement mis en œuvre, il est nécessaire de traquer les cellules résiduelles ou réémergentes en utilisant toute une panoplie de thérapies qui s’adresseront, de façon de plus en plus spécifique et personnalisée, à chaque type de tumeur et à chaque patient. Le petit convoyeur TAT et les molécules ciblant des domaines définis du p53 muté ne seront pas les derniers à remplir ce rôle.
Certes, les résultats ne sont pas instantanément transférables de la souris à l’Homme, et certes les mutations s’accumulent avec l’allongement de la vie. Mais les traitements des cancers, au long d’un parcours jalonné de succès et d’échecs, se font de plus en plus précis, et sur des fronts de plus en plus variés.
Bibliographie
-
Page de définition du cancer du site de l’Institut National du Cancer.
-
Oncogenes and Tumor Suppressor Genes . Site de l’American Cancer Society.
-
Kashiwagi H. et coll. TAT-Bim Induces Extensive Apoptosis in Cancer Cells . Annals of surgical oncology. (2007) 14:1763-1771.
-
Ericson G. HIV protein enlisted to help kill cancer cells. Site EurekAlert ! (2007).
-
Une molécule au cœur des mécanismes du cancer. La recherche (1999) 323.
-
Denis Herman Les différents types de sénescence et de mort cellulaires. Dans Sénescence et mort cellulaires. Causes, origine et remèdes.
-
Wen Xue W. et coll. Senescence and tumour clearance is triggered by p53 restoration un murine liver carcinomas (texte complet). Nature (2007) 445:656-660.
-
Ventura A. et coll. Restoration of p53 function leads to tumour regression in vivo. Nature (2007) 445:661-665. (texte complet)
-
Ibarrondo F. Une nouvelle classe d’ARN : les petites ARN interférents. Site « planet-vie »
-
Martins C.P., Brown-Swigart L. et Evan G.I. Modeling the Therapeutic Efficacy of p53 Restoration in Tumors (texte complet). Cell (2006) 127:1323-1334.
-
Perkel J.M. Tumors shrink when p53 restored . The new scientist (2007)
-
Bouchet B.P., Caron de Fromentel C., María Galmarini C. et Puisieux A. p53 comme cible thérapeutique pour le développement de médicaments anticancéreux (texte complet). Bulletin du Cancer (2006) 93:145-153.