En réponse aux agressions de l’environnement, les cellules de notre organisme entrent parfois dans un état appelé sénescence cellulaire. Longtemps considérée comme étant uniquement à l’origine du vieillissement de l’organisme, de nombreuses études scientifiques ont aujourd’hui établi le rôle déterminant que cette destinée cellulaire joue dans le développement embryonnaire, la cicatrisation ou encore comme barrière à l’émergence de cancers.
La sénescence est un état cellulaire caractérisé par un arrêt irréversible du cycle cellulaire, ainsi que par des modifications cellulaires et moléculaires particulières. Elle fut décrite pour la première fois dans les années 1960 lorsque Hayflick et Moorheads démontrèrent que les capacités de prolifération de fibroblastes humains in vitro étaient limitées [1]. Au cours de leurs expérimentations, ils observaient qu’à l’issue d’un nombre défini de divisions cellulaires, le nombre de mitoses observées déclinait pour finalement devenir nul, et ce malgré l’apport de facteurs de croissance dans le milieu de culture et l’absence d’inhibition de contact.
Peu après cette découverte, deux hypothèses majeures – en apparence contradictoires – virent le jour pour tenter d’expliquer que les cellules normales ne prolifèrent pas indéfiniment. La première hypothèse reposait sur le fait que de nombreuses cellules tumorales disposent d’une capacité de prolifération illimitée in vitro et plaçait par conséquent la sénescence cellulaire au rang de potentiel mécanisme suppresseur de tumeurs, protégeant l’organisme contre l’émergence de cancers. Cependant, conscients du déclin des capacités de régénération tissulaire observé avec l’avancée en âge chez les espèces mammaliennes, de nombreux scientifiques percevaient alors la sénescence cellulaire comme un mécanisme largement délétère in vivo et suspectaient qu’elle participe à l’émergence de pathologies liées à l’âge.
Ces deux hypothèses furent explorées plus ou moins indépendamment durant de nombreuses années, et – bien que paradoxales – elles sont à l’heure actuelle toutes deux éprouvées et constituent les piliers de la recherche contemporaine sur la sénescence cellulaire. En effet, de nombreuses études ont établi l’implication physiologique significative de ce mécanisme dans le développement embryonnaire, la réparation tissulaire ou encore la suppression de tumeurs. Mais en dépit de son effet bénéfique pour l’organisme, de multiples travaux montrent que, dans sa composante chronique, la sénescence constitue un processus nuisible potentiellement tumorigénique et pouvant altérer l’homéostasie tissulaire, favorisant ainsi l’émergence de pathologies neurodégénératives, inflammatoires ou encore métaboliques (Figure 1).

La sénescence cellulaire est plus qu’un programme antiprolifératif. Les cellules sénescentes sécrètent des facteurs qui constituent le phénotype sécrétoire pro-inflammatoire associé à la sénescence. En général, par le biais des molécules pro-inflammatoires qu’elles sécrètent, les cellules sénescentes recrutent les acteurs de l’immunité qui procèdent alors à leur élimination. En plus de ses fonctions dans le développement embryonnaire ou la cicatrisation, la sénescence cellulaire constitue une réelle barrière au développement tumoral. Cependant, si leur élimination n’a pas lieu, les cellules sénescentes s’accumulent, favorisant le développement de cancers et la survenue de pathologies liées à l’âge. Différentes stratégies thérapeutiques (en rouge) peuvent être utilisées pour exploiter les aspects bénéfiques de la sénescence cellulaire et en réprimer les aspects négatifs. Par exemple, les molécules sénomorphes induisent la sénescence tandis que les molécules sénolytiques détruisent les cellules dans cet état. Adapté d’après [2].
Cet article offre un tour d’horizon des connaissances scientifiques actuelles relatives à la sénescence cellulaire. Après avoir établi la carte d’identité phénotypique des cellules sénescentes (i) et évoqué ses origines évolutives (ii), nous verrons qu’il s’agit en réalité d’une destinée cellulaire plurielle pouvant être induite par des stimuli de natures variées et impliquant différentes voies de signalisation cellulaire (iii et iv). À la lumière des travaux de recherche les plus récents, nous développerons ses implications physiologiques (vi) et ses conséquences pathophysiologiques in vivo (vii), en particulier quant à son rôle dans la promotion et la suppression tumorale et le vieillissement, tout en évoquant les perspectives thérapeutiques émergentes reposant sur les molécules sénomorphes, qui induisent la sénescence, et sénolytiques, qui détruisent les cellules sénescentes.
À la recherche de biomarqueurs spécifiques
En fonction du type cellulaire et du contexte biologique, les cellules de mammifères exposées à certains stress cellulaires peuvent entrer en sénescence. Celle-ci se traduit par des modifications cellulaires et l’expression de protéines particulières (voir infra) et également par un arrêt de prolifération classiquement considéré comme irréversible en phase G1, laissant les cellules dans un état métabolique et transcriptionnel actif. Cette transition vers la sénescence s’accompagne le plus souvent d’une résistance aux signaux apoptotiques, d’une augmentation de l’activité de l’enzyme β-galactosidase lysosomiale, ainsi que de profondes modifications cytomorphologiques (aplatissement cellulaire, vacuolisation du cytoplasme, augmentation du volume nucléaire) et des altérations majeures de l’architecture chromatinienne et de l’expression génique (Figure 2).
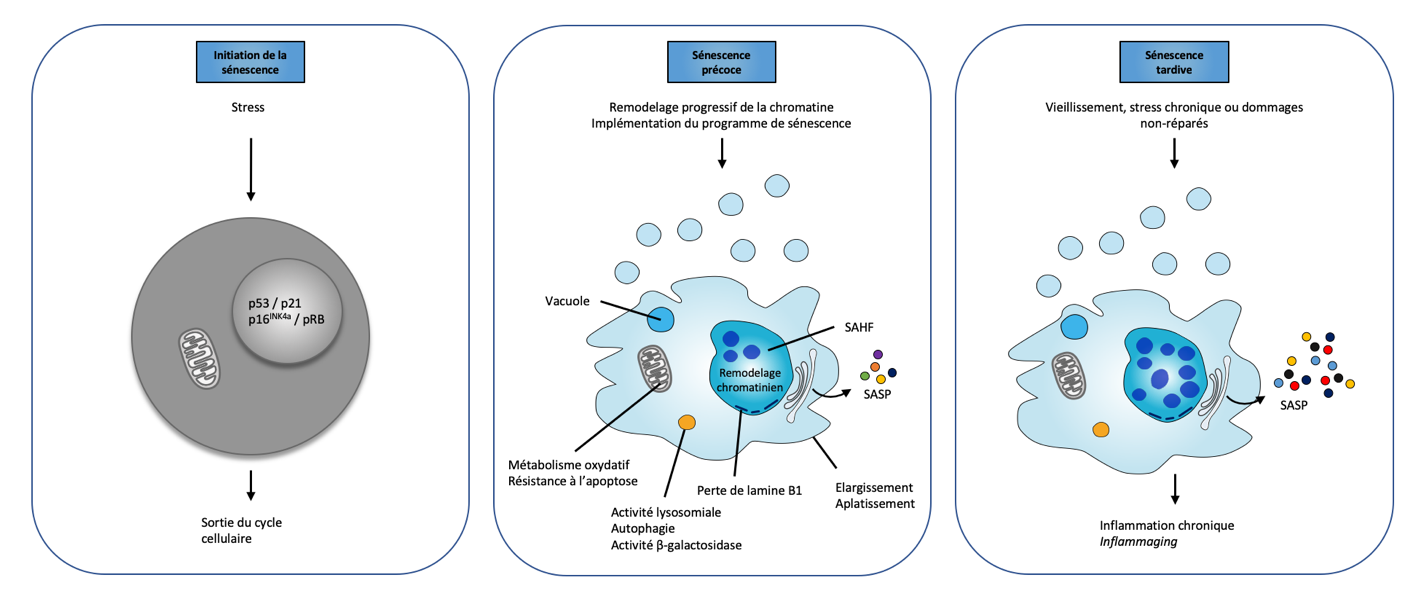
Sur ces schémas sont représentés les principales altérations qui émergent au niveau cellulaire au cours des différentes phases de la sénescence. Lors de la phase initiale, qui se matérialise par la sortie du cycle cellulaire, on observe essentiellement une activation des protéines p53, p21, p16INK4a et pRB. Lors de la phase précoce, émergent les modifications cytomorphologiques (aplatissement et donc élargissement de la cellule), les modifications chromatiniennes (nucléations des foyers d’hétérochromatine associés à la sénescence (SAHF), perte de lamine B1), alors que l’activité de la β-galactosidase lysosomiale augmente de manière significative. À cette étape, la cellule sécrète déjà un grand nombre de molécules pro-inflammatoires (SASP). Lorsque la sénescence persiste, les modifications cytomorphologiques et chromatiniennes s’accentuent et la nature du SASP évolue, favorisant l’inflammation chronique. Adapté d’après [3].
La sénescence est un état cellulaire initié par un stress et dont l’étape première est l’arrêt du cycle cellulaire en phase G1. S’ensuit alors une modification progressive de l’architecture chromatinienne initiée par une diminution de l’expression de la lamine B – permettant en temps normal la fixation de l’ADN au niveau de la membrane nucléaire – qui se matérialise par l’émergence des foyers d’hétérochromatine associés à la sénescence (SAHF, senescence-associated heterochromatin foci). Ces derniers sont caractérisés par les marqueurs d’hétérochromatine transcriptionnellement réprimée (p. ex. di et triméthylation sur la lysine 9 de l’histone H3 : H3K9me2/3).
Ces modifications de la chromatine ont pour conséquences de bloquer de façon permanente les cellules dans un état sénescent en réprimant les gènes impliqués dans le cycle cellulaire tout en favorisant l’expression progressive des gènes codant les protéines constitutives du phénotype sécrétoire associé à la sénescence (SASP, senescence-associated secretory phenotype). Ce sécrétome spécifique associant interleukines (par exemple IL1α, IL1β, IL6), chimiokines (p. ex. IL8, INFɣ), protéases (MMP1, MMP3) et facteurs de croissance (p. ex. EGF, VEGF, HGF) – qui agit aussi bien en paracrine qu’en autocrine – joue un rôle important dans la réponse inflammatoire, le remodelage de la matrice extracellulaire et le maintien de l’état sénescent. Aussi, en phase précoce, il assure le recrutement du système immunitaire permettant l’élimination des cellules sénescentes. En phase tardive cependant et lorsque les cellules sénescentes s’accumulent, il génère une inflammation chronique – parfois appelée inflammaging – contribuant à la déplétion du réservoir de cellules souches, altérant l’homéostasie tissulaire et pouvant favoriser la transformation des cellules du microenvironnement en cellules pro-tumorales.
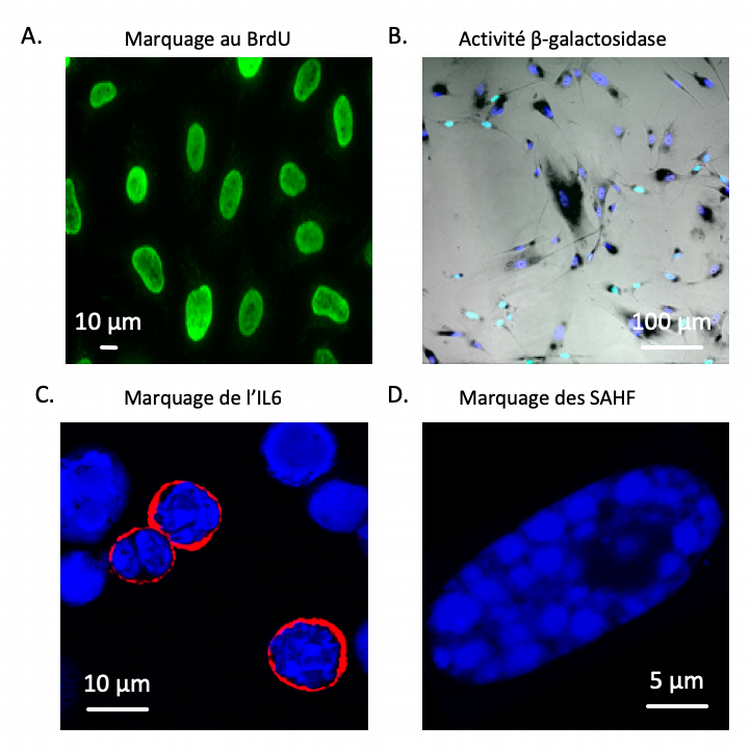
Les marqueurs présentés ici, bien que non spécifiques de la sénescence lorsque considérés isolément, sont souvent utilisés en combinaison. A. Image d’immunofluorescence montrant des cellules proliférantes marquées au BrdU (5-bromo-2’-désoxyuridine), un analogue synthétique de la thymidine qui est incorporé dans l’ADN durant la phase S du cycle cellulaire et qui ne peut être incorporé par les cellules ayant cessé de se diviser. B. Superposition d’images montrant des fibroblastes humains sénescents in vitro. Les noyaux cellulaires sont marqués en bleu foncé (coloration au DAPI (4’,6-diamidino-2-phénylindole), une molécule fluorescente capable de se lier fortement aux bases adénine et thymine de l’ADN). Le bleu clair marque les noyaux ayant incorporé du BrdU. Enfin, la coloration noire reflète l’activité β-galactosidase lysosomiale C. Image d’immunofluorescence montrant les noyaux cellulaires en bleu (coloration au DAPI) et la protéine IL6 en rouge. D. Image d’immunofluorescence montrant un unique noyau cellulaire (coloration au DAPI) présentant de nombreux SAHF.
En substance, l’arrêt du cycle cellulaire est une caractéristique cruciale pour l’identification de tous les types de sénescence, mais il ne peut être considéré comme un marqueur biologique de la sénescence puisque que de multiples mécanismes cellulaires peuvent entraîner un arrêt réplicatif stable. Cependant, l’incapacité des cellules sénescentes à ré-exprimer les gènes nécessaires à la prolifération, même dans un environnement pro-mitogène, permet de distinguer la sénescence de la quiescence – un état non prolifératif des cellules qui s’inverse facilement en réponse aux mitogènes. Au-delà de l’arrêt réplicatif stable, nous avons vu que le phénotype sénescent comporte d’autres éléments caractéristiques. Cependant, les changements énumérés ci-dessus ne se matérialisent pas tous simultanément dans l’ensemble des types cellulaires et il existe, par ailleurs, une grande hétérogénéité phénotypique en fonction du stimulus impliqué. En réalité, il n’existe à l’heure actuelle pas de biomarqueur complètement spécifique à cette destinée cellulaire, et l’on utilise aujourd’hui encore une combinaison d’indicateurs qui lui sont associés – tels qu’un marquage reflétant l’activité β-galactosidase lysosomiale, un autre permettant de visualiser les SAHF, un autre encore matérialisant l’arrêt de division cellulaire, et la mesure d’expression de gènes codant des protéines du phénotype sécrétoire associé à la sénescence (SASP) – pour mettre en évidence les cellules sénescentes in vitro et in vivo (Figure 3). Aussi, de nombreuses équipes de recherche tentent à l’heure actuelle d’identifier des biomarqueurs de la sénescence qui feraient consensus pour l’ensemble des types cellulaire possiblement impactés et l’ensemble des stimuli pouvant l’induire.
Comme nous avons pu le voir ici, malgré un arrêt réplicatif stable, les cellules sénescentes restent métaboliquement actives et sont capables de produire et de sécréter une pléthore de facteurs pouvant affecter le microenvironnement tissulaire selon différentes modalités. Nous verrons dans la suite de cet article que ce sécrétome particulier est en grande partie à l’origine des implications physiologiques et physiopathologiques de la sénescence cellulaire dans l’organisme.
Origine évolutive de la sénescence cellulaire
Il est aujourd’hui bien établi que la sénescence est l’une des causes principales du vieillissement et des pathologies liées l’âge. Aussi, pendant de nombreuses années, les scientifiques se sont demandés pourquoi la capacité de sénescence n’avait pas été contre-sélectionnée. Les récentes études mettant en exergue son rôle déterminant dans la régulation de l’embryogenèse ainsi que son action antitumorale contribuent à expliquer son maintien à l’échelle évolutive. En effet, ces conjectures sont cohérentes avec l’hypothèse de la pléiotropie antagoniste, selon laquelle les allèles pouvant avoir un effet délétère chez un individu âgé ne sont pas nécessairement contre-sélectionnés s’ils ont des effets bénéfiques tôt dans la vie.
À l’échelle du règne animal, il est intéressant de noter que plusieurs espèces ne présentent pas de sénescence cellulaire. Les travaux du biogérontologiste Caleb Finch établissent ainsi que, chez certaines espèces d’esturgeons, de tortues, de mollusques, de crustacés et de cnidaires, les individus peuvent vivre jusqu’à un âge considérable dans la nature sans que leur succès reproducteur ne soit impacté, ni que des signes de sénescence cellulaire (p. ex. érosion télomérique, dommages à l’ADN accrus) ne soient observés [4]. Chez l’hydrozoaire Turritopsis nutricula, cette absence de sénescence cellulaire au cours de la vie s’accompagne de la capacité singulière du stade adulte et sexuellement mature (stade méduse) à retourner à l’état juvénile (stade polype). Il est important de noter que ces animaux ne sont pas sujets au développement de cancers et que l’embryogenèse et le remodelage tissulaire sont régulés selon des modalités différentes de celles des mammifères.
Aujourd’hui, de nombreuses équipes de recherche tentent de comprendre les raisons évolutives justifiant l’absence de sénescence cellulaire chez ces espèces métazoaires, ou encore de décrypter les processus biologiques impliqués dans le cycle de vie singulier de Turritopsis nutricula, dans l’optique d’élucider les forces évolutives à l’œuvre au cours de la vie des organismes vivants, mais également d’identifier de potentielles cibles thérapeutiques pour le développement de molécules sénolytiques ou sénomorphes.
Une destinée cellulaire aux origines multiples
La sénescence cellulaire fut à l’origine identifiée comme phénomène consécutif au raccourcissement télomérique lié aux mitoses successives des cellules en réplication. Depuis, de nombreuses études ont établi qu’au-delà de cette sénescence réplicative (RS, replicative senescence) existait une panoplie de destinées cellulaires regroupées sous le terme de sénescence prématurée induite par le stress (SIPS, stress-induced premature senescence), provoquée par des stimuli variés tels que les stress génotoxiques, le stress oxydatif, l’hyperactivation d’oncogènes ou encore les dommages tissulaires, et pouvant également impacter les cellules postmitotiques.
Érosion télomérique
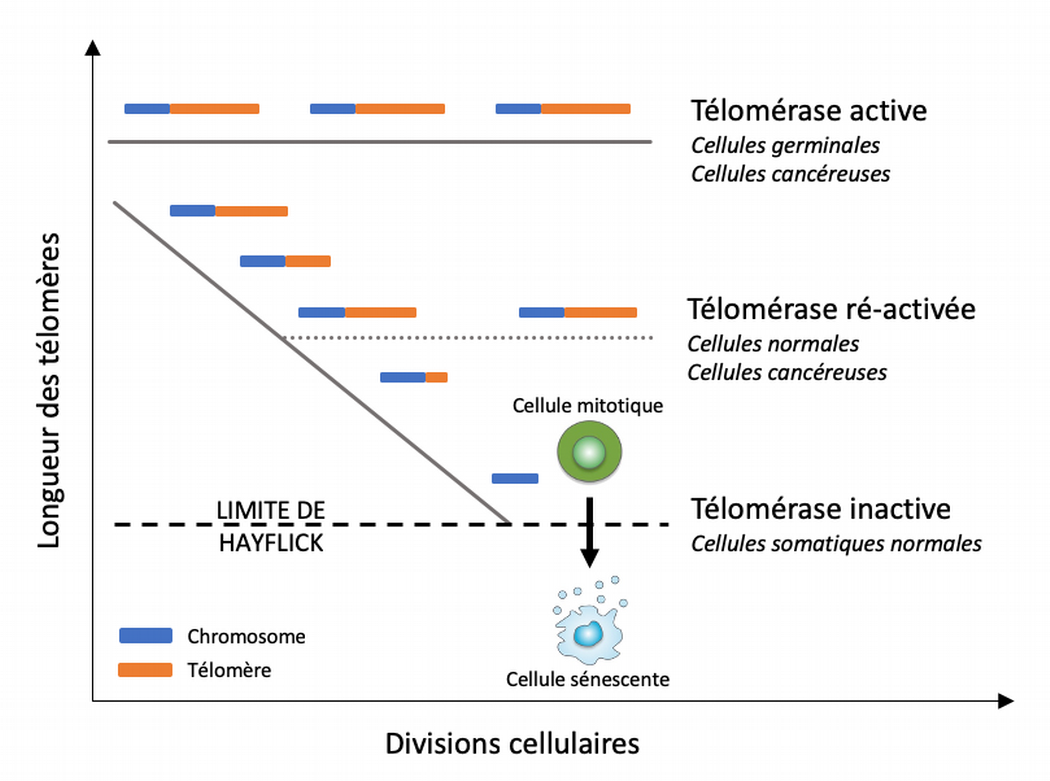
La longueur des télomères est maintenue par l’enzyme télomérase, qui est exprimée par les cellules qui composent la lignée germinale ainsi que par de nombreuses cellules cancéreuses. La plupart des cellules somatiques normales n’expriment pas cette enzyme, ou ne l’expriment que de façon transitoire ou à des niveaux trop faibles pour empêcher le raccourcissement des télomères causé par les divisions cellulaires successives. Aussi, la longueur des télomères diminue à chaque cycle cellulaire. Une fois la limite de Hayflick atteinte, les télomères devenus dysfonctionnels déclenchent une réponse aux dommages à l’ADN, auquel les cellules réagissent en entrant en sénescence. Modifié d’après [5].
Chez les mammifères, les télomères correspondent à des séquences d’ADN répétées (5’-TTAGGG-3’ chez les vertébrés) associées au complexe protéique shelterin et l’ARN long non codant TERRA formant le capping des chromosomes linéaires, les protégeant de potentielles dégradations ou fusions au cours des processus de réparation de l’ADN. Bien que l’architecture télomérique mammalienne ne soit pas encore parfaitement établie, on sait qu’ils se terminent par une structure en boucle nommée t-loop. Pour des raisons biochimiques, l’ADN polymérase – qui assure la copie du matériel génétique lors de la division cellulaire – ne peut garantir la réplication de l’entièreté de ces extrémités. Aussi, au cours de chaque cycle réplicatif et en l’absence d’activité télomérase – une enzyme assurant la réparation des télomères, essentiellement active dans les cellules germinales et cancéreuses – les télomères raccourcissent de 50 à 200 bp (Figure 4) jusqu’à atteindre la limite de Hayflick, au-delà de laquelle ils déclenchent alors le signal de sénescence médié par les voies de signalisation de la réponse aux dommages à l’ADN (DDR, DNA damage response).
Dommage à l’ADN et stress oxydatif
La cellule dans son environnement se voit par ailleurs perpétuellement confrontée à de nombreux stress qui menacent son intégrité génomique, tels que les rayonnements ultraviolets ou les radiations ionisantes, alors que le métabolisme cellulaire basal produit lui-même des espèces réactives de l’oxygène (ROS) aux propriétés génotoxiques. Par ailleurs, les polymérases qui assurent la réplication de l’ADN lors de la mitose commettent parfois des erreurs qui rompent la complémentarité entre les brins de la double hélice. Aussi, on estime que dans un organisme mammalien donné émergent de 1 000 à 1 000 000 de lésions de l’ADN par cellule et par jour, qu’il s’agisse de coupures simple brin (SSB, single-strand break), de cassures double brin (DSB, double-strand break), d’altérations de base ou encore de liens inter/intra-brins.
La réponse aux dommages à l’ADN (DNA damage response, DDR) implique un réseau de signalisation complexe et intriqué assurant la détection de l’ADN endommagé, l’arrêt de la progression du cycle cellulaire et la mobilisation des acteurs nécessaires à la réparation lorsque celle-ci est envisageable (Figure 5). Bien que l’étendue des dommages semble déterminante, on sait peu de choses sur les raisons pour lesquelles les cellules présentent soit une activation transitoire de la DDR – qui mène à un arrêt réversible de la division cellulaire le temps des réparations – soit une signalisation persistante de la DDR – qui aboutira à la sénescence cellulaire. On sait cependant que la voie signalétique empruntée par la réponse aux dommages à l’ADN est spécifique au type de lésions et qu’elle est déterminée par la reconnaissance des dommages par les complexes protéiques senseurs (complexe MRE11-RAD50-NBS50 pour les DSB, complexe RAD9-RAD1-HUS1 pour les SSB), le recrutement de protéines médiatrices (53BP1, H2AX, BRCA1), puis par l’activation des PI3K-like kinases ATM (ataxia-telangectasia mutated), ATR (ataxia-telangectasia and Rad3-related) et de DNA-PKcs (DNA-dependent protein kinase catalytic subunit) qui phosphorylent et activent par la suite diverses protéines effectrices coordonnant l’arrêt du cycle cellulaire et, éventuellement, la réparation des dommages ou l’entrée en sénescence cellulaire.
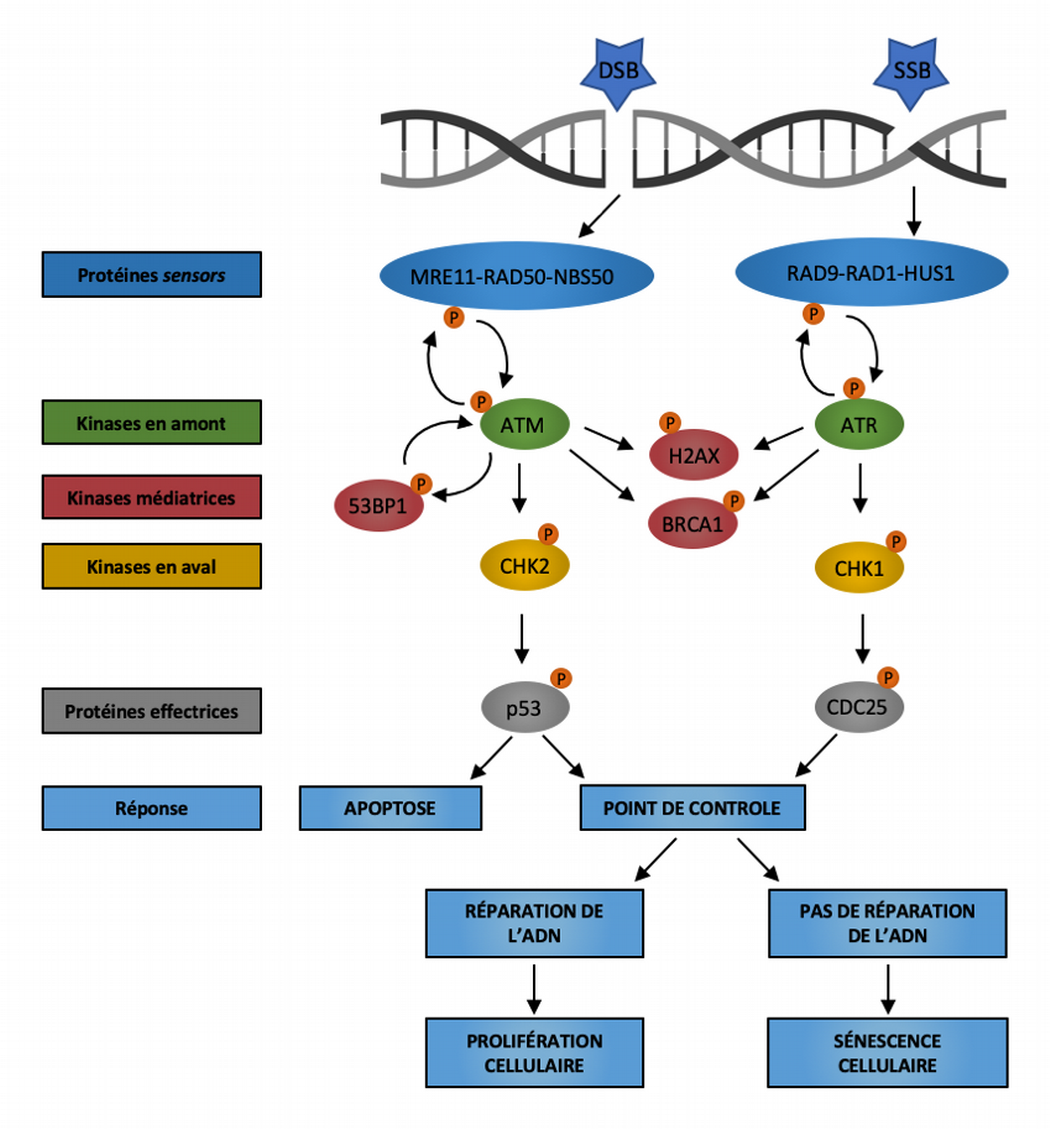
La voie de réponse aux dommages à l’ADN (DDR) repose sur deux principaux complexes protéiques senseurs détectant les dommages à l’ADN : le complexe MRE11-RAD50-NBS1 qui détecte les cassures double brin (DSB) et le complexe RAD9-RAD1-HUS1 qui détecte les régions exposées d’ADN monocaténaire (SSB). Ces senseurs recrutent alors les kinases ATM et ATR qui, à leur tour, phosphorylent le variant d’histone H2AX dans la région proximale de la lésion de l’ADN. Alors qu’ATM est principalement activée par les cassures double brin, ATR est essentiellement induite par les cassures simple brin causées par la réplication de l’ADN et l’hyperactivation d’oncogènes. La protéine BRCA1 est recrutée sur les sites d’altération de l’ADN après phosphorylation par ATM et ATR. La protéine 53BP1 participe également au maintien de la signalisation de réponse aux dommages de l’ADN en favorisant l’activation d’ATM. La suite de la signalisation DDR se déroule à distance du locus endommagé via le recrutement des kinases diffusibles CHK2 et CHK1, et converge vers des effecteurs en aval tels que la phosphatase CDC25 et le facteur de transcription p53. Les conséquences de l’activation de la réponse aux dommages de l’ADN peuvent être la mort cellulaire par apoptose, l’arrêt transitoire du cycle cellulaire suivi de la réparation des dommages à l’ADN et de la reprise de la prolifération, ou la sénescence cellulaire causée par la persistance des dommages non réparés de l’ADN. Modifié d’après [6].
Hyperactivation d’oncogènes
Les oncogènes sont des versions mutantes de gènes normaux, initialement impliqués dans la stimulation du cycle cellulaire ou dans l’inhibition de l’apoptose, et ayant acquis le potentiel d’induire une prolifération anarchique des cellules et la formation de néoplasies. En 1997, Scott Lowe et son équipe montraient que lorsqu’une forme oncogène de RAS – un transducteur cytoplasmique de signaux mitogènes – était exprimée dans des fibroblastes humains sains, ces derniers entraient en sénescence cellulaire, soulignant pour la première fois le rôle onco-suppressif joué par la sénescence cellulaire in vitro [7]. Par la suite, d’autres membres de la voie de signalisation de RAS (p. ex. RAF, MEK, MOS et BRAF), ainsi que des protéines nucléaires pro-prolifératives (p. ex. E2F1), se sont révélés induire la sénescence lorsqu’ils étaient surexprimés.
Quelques années à peine après la découverte de Lowe et al., des études venaient attester de l’existence de la sénescence induite par oncogènes (OIS, oncogene-induced senescence) in vivo chez l’humain et la souris, en réponse à divers oncogènes et dans des contextes histologiques variés (p. ex. tumeur mammaire, adénome pulmonaire, carcinome hépatocellulaire, sarcome). On sait aujourd’hui, par exemple, que les foyers de tumeurs bénignes mélanocytaires (naevi, communément appelées « grains de beauté ») sont occupés par des cellules devenues sénescentes en réponse aux oncogènes BRAF ou NRAS, empêchant leur évolution vers des lésions malignes.
Les fondements moléculaires de la sénescence
Qu’elle soit initiée par un stress génotoxique, l’érosion télomérique, ou l’hyperactivation d’un oncogène, l’établissement de la sénescence cellulaire repose sur deux piliers fondamentaux : l’arrêt quasi irréversible du cycle cellulaire en phase G1 et le changement progressif du programme transcriptionnel.
En 1991, les travaux de Shay et Woodring [8] révélaient le rôle essentiel que jouent les protéines p53 (tumor protein 53, surnommée « le gardien du génome ») et pRB (retinoblastoma protein) dans l’établissement de la sénescence. Caractérisées quelques années auparavant (en 1979 et 1986 respectivement) dans le cadre de recherches sur le cancer, ces deux suppresseurs de tumeurs sont aujourd’hui connus pour leur rôle déterminant dans la régulation du cycle cellulaire, l’apoptose, l’autophagie et l’entrée en sénescence.
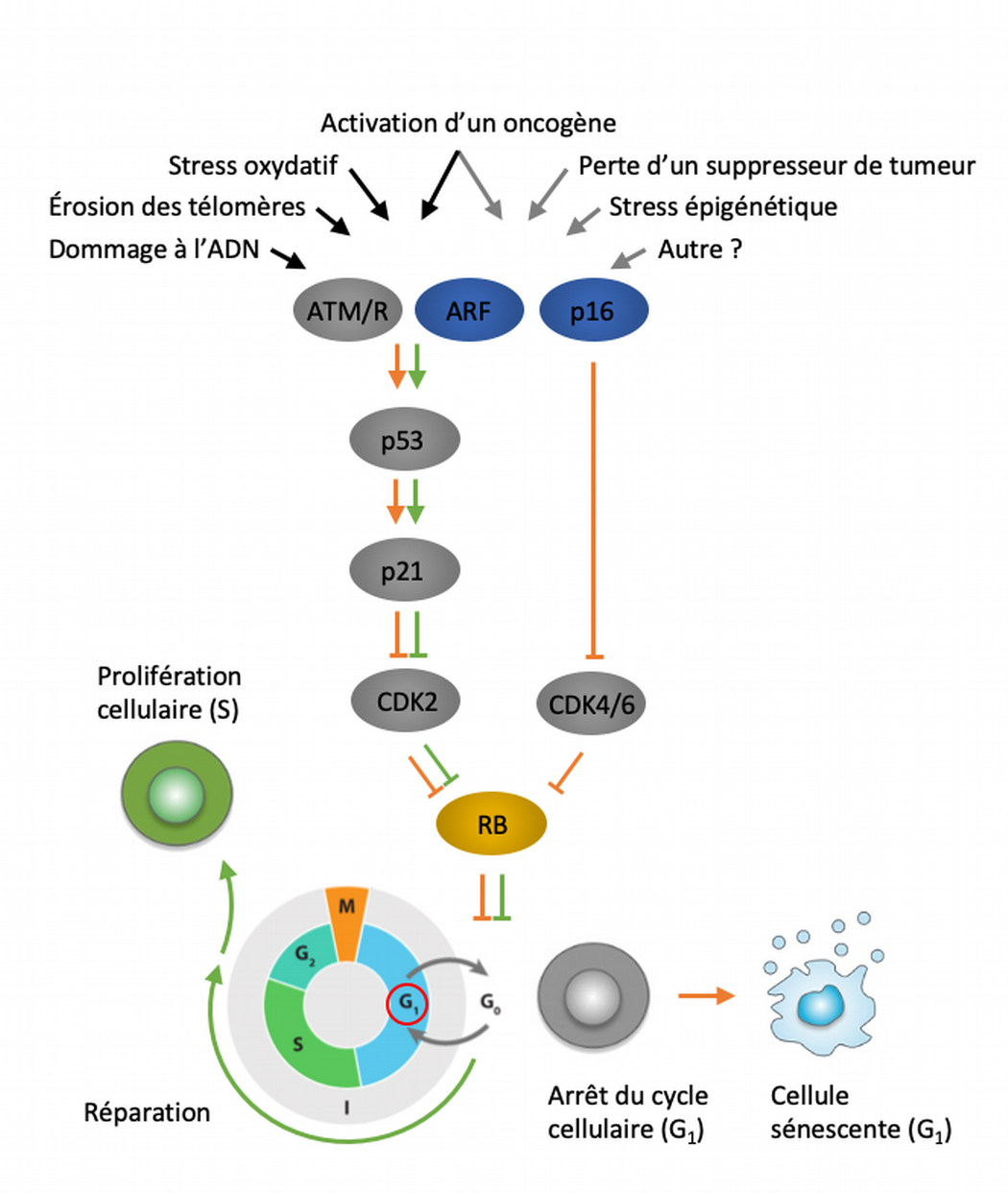
Une multitude de stimuli peuvent activer le programme de sénescence cellulaire. Ces facteurs de stress déclenchent diverses cascades de signalisation cellulaire qui convergent vers p53 et p16Ink4a. Les types de stress qui activent p53 via la signalisation de réponse aux dommages de l’ADN sont indiqués par des flèches noires. L’activation de p53 induit p21, qui induit un arrêt réversible du cycle cellulaire en inhibant la cycline CDK2. p16Ink4a inhibe également la progression du cycle cellulaire mais le fait en ciblant les complexes cyclines CDK4/CDK6. À terme, p21 et p16Ink4a entravent l’inactivation de pRB, ce qui entraîne une répression continue des gènes cibles E2F nécessaires à la transition vers la phase S. En cas de stress majeur (flèches orange), les cellules entrent en sénescence via un mécanisme encore mal compris. Les cellules exposées à des dommages superficiels pouvant être réparés avec succès peuvent reprendre une progression normale du cycle cellulaire (flèches vertes). Modifié d’après [9].
Quel que soit le facteur de stress impliqué dans l’initiation de la sénescence cellulaire, les cascades de signalisation cellulaire finissent par activer les protéines p53 ou p16Ink4a (Figure 6). Pour la grande majorité des sources de stress, par un jeu de phosphorylations successives, l’activation de p53 induit p21, qui induit un arrêt temporaire du cycle cellulaire en inhibant la kinase cycline-dépendante CDK2. Lorsque la sénescence est induite par l’hyperactivation d’un oncogène ou la perte d’activité d’un suppresseur de tumeur, une seconde voie signalétique est mobilisée, impliquant l’inhibition des kinases cycline-dépendantes CDK4 et CDK6 via p16Ink4a. Dans les deux situations, les kinases cycline-dépendantes ne peuvent assurer l’inactivation de pRB par déphosphorylation, entraînant de fait une répression continue des gènes cibles des facteurs de transcription E2F qui sont indispensables à la transition vers la phase S du cycle cellulaire. Aussi, ces deux voies signalétiques, qui agissent potentiellement en synergie, convergent pour bloquer les cellules lésées en phase G1.
Les cellules exposées à des dommages superficiels qui peuvent être réparés avec succès pourront à terme entrer à nouveau dans le cycle cellulaire. En revanche, en cas de stress majeur, les cellules entament leur entrée en sénescence par un mécanisme encore mal appréhendé, mais caractérisé par un arrêt quasi irréversible en phase G1.
Le stress oxydatif généré par l’activité mitochondriale de la cellule entrée en sénescence va stimuler de manière concomitante l’activité de la MAP kinase p38, induisant à son tour l’activation du facteur de transcription NFκB. En coordination avec le facteur de transcription CEBPβ, il va progressivement assurer l’expression des gènes de la réponse inflammatoire et à l’établissement du phénotype sécrétoire associé à la sénescence.
La sénescence dans le remodelage tissulaire
Le programme de sénescence est impliqué dans de nombreux processus physiologiques et pathologiques qui reposent sur le remodelage tissulaire. Dans ce contexte, c’est le degré de persistance des cellules sénescentes au cours de ces processus qui déterminera si elles y jouent un rôle positif ou négatif : leur présence transitoire dans les tissus couvre principalement des fonctions bénéfiques, alors que leur accumulation aura un impact négatif sur la restauration de l’homéostasie tissulaire (Figure 7).
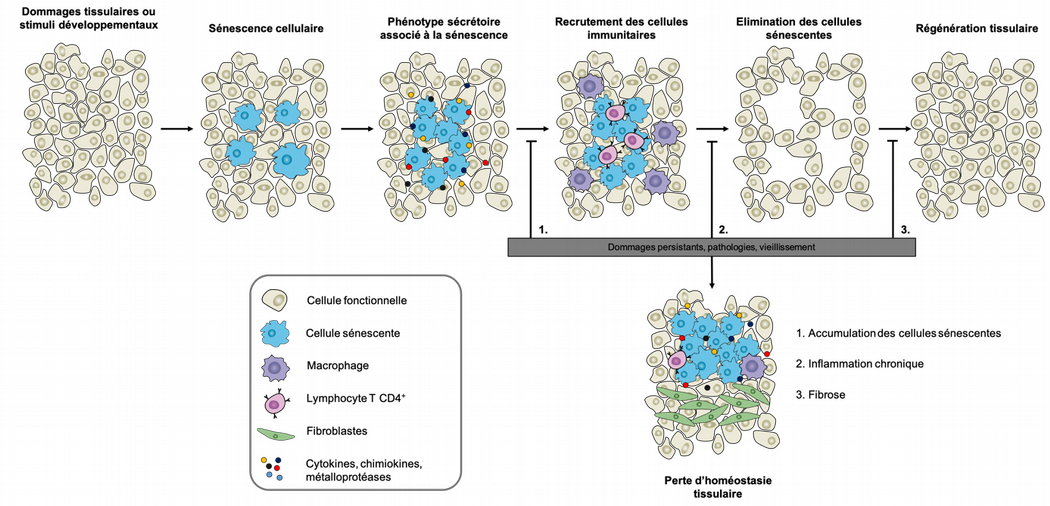
La sénescence amorce un processus de remodelage tissulaire en recrutant des cellules immunitaires par le biais de molécules sécrétées (cytokines et chimiokines). Les macrophages éliminent les cellules sénescentes et les cellules progénitrices repeuplent et régénèrent les tissus endommagés. Cette séquence sénescence-élimination - régénération peut être altérée en cas de lésions persistantes, de situations pathologiques ou de vieillissement. Dans ces cas, les cellules sénescentes ne sont pas éliminées efficacement et les tissus ne sont pas entièrement régénérés. Modifié d’après [10].
Au cours de l’embryogenèse
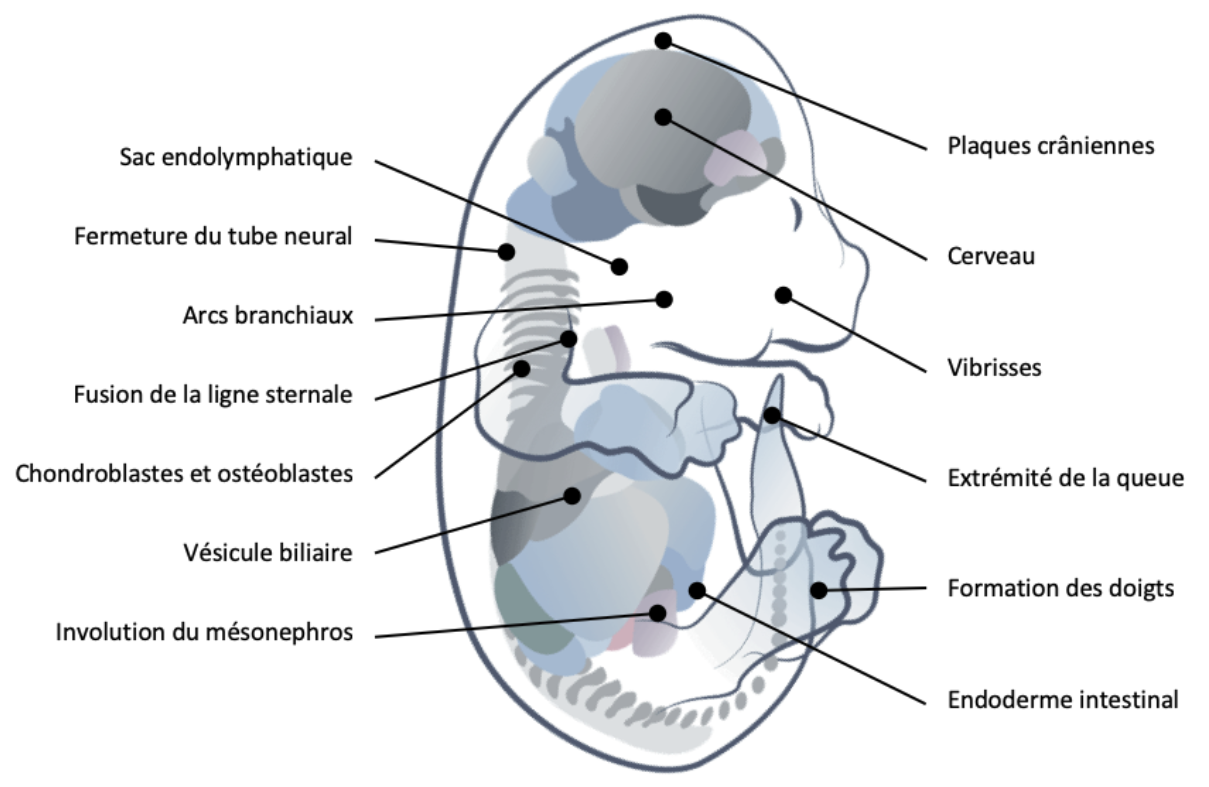
Régions de l’embryon de souris au sein desquelles ont été observées des cellules positives à des marqueurs de sénescence cellulaire. Modifié d’après [10].
Les recherches sur la sénescence se sont, dans un premier temps, surtout focalisées sur la compréhension des mécanismes impliqués dans son émergence dans un contexte physiopathologique. Mais en 2013, les équipes de Manuel Serrano et de William Keyes bouleversaient les dogmes alors établis en révélant l’identification de cellules sénescentes dans un grand nombre de structures embryonnaires chez la souris et le poulet [11, 12]. Ultérieurement, ces observations furent étendues à l’être humain, suggérant de fait que cette caractéristique est conservée à l’échelle de l’évolution des vertébrés. On sait aujourd’hui que la sénescence cellulaire intervient, au cours de l’embryogenèse des vertébrés, dans l’involution du mésonéphros au niveau rénal, la formation du sac endolymphatique de l’oreille interne, la régression des palmes interdigitales ou encore la fermeture du tube neural.
Au cours de la cicatrisation et de la régénération
En réponse à une lésion tissulaire ou à une blessure, des mécanismes sophistiqués existent chez les mammifères pour prévenir les infections par des agents pathogènes étrangers et pour réparer les tissus endommagés. La réparation tissulaire est un phénomène reposant sur les phases d’hémostase, d’inflammation, de prolifération et de remodelage, toutes fortement influencées par les cellules sénescentes via le phénotype sécrétoire associé à la sénescence.
Ainsi, en 2014, Judith Campisi et son équipe démontraient que l’induction d’une plaie chez la souris entraînait l’apparition transitoire de fibroblastes sénescents sécrétant des cytokines assurant le recrutement de cellules endothéliales au site de la lésion [13]. En utilisant un modèle murin génétiquement modifié dans lequel les cellules sénescentes pouvaient être éliminées à la demande, ils montraient par ailleurs que l’élimination de ces cellules retardait le processus de cicatrisation. Au cours d’expérimentations plus approfondies, ils mettaient enfin en évidence que les cellules sénescentes localisées au niveau de la lésion secrètent le facteur de croissance PDGF-AA induisant la différenciation des fibroblastes voisins en myofibroblastes qui en se contractant permettent de refermer la plaie, optimisant ainsi la cicatrisation.
Par ailleurs, les formidables capacités de régénération des urodèles sont également en lien avec la sénescence cellulaire. Aussi, chez la salamandre, après amputation d’un membre, les cellules sénescentes apparaissent de façon transitoire pendant les stades intermédiaires de régénération avant d’être éliminées par les macrophages au cours de la phase de maturation [14]. On ne sait pas encore le rôle exact que la sénescence joue dans ce processus mais elle est probablement à l’origine, via le phénotype sécrétoire associé à la sénescence, du recrutement des macrophages assurant le remodelage tissulaire, comme c’est le cas lors de l’embryogenèse.
Au cours de la fibrose
En réponse à un dommage tissulaire ou à un phénomène inflammatoire, se met en place une phase de réparation tissulaire pouvant entraîner la formation d’un excès de tissu conjonctif : c’est la fibrose. En situation physiologique, l’accumulation de protéines de la matrice extracellulaire (p. ex. collagènes de type I et III, fibronectine, laminine) assure la cicatrisation permanente. Néanmoins, ce processus affecte profondément la structure et la fonctionnalité des tissus et peut, dans certains cas, entraîner la défaillance des organes. Dans ce contexte, il a été démontré que la sénescence cellulaire interférait dans la formation des tissus cicatriciels.
Aussi, lors d’une blessure au niveau cutané et au cours de la phase de remodelage de la lésion cicatrisée, la protéine matricellulaire CYR61 (cysteine-rich angiogenic inducer 61, aussi appelée CCN1) induit la sénescence des fibroblastes dans le microenvironnement. Elle active dans un premier temps la NADPH oxydase 1 (NOX1) par l’intermédiaire de RAC1 (Ras-related C3 botulinum toxin substrate 1). En résulte une augmentation significative de la production d’espèces réactives de l’oxygène, qui activent p53 via la réponse aux dommages de l’ADN et p16 via la MAP kinase p38. En réponse, les fibroblastes devenus sénescents sécrètent des métalloprotéases matricielles antifibrotiques qui assurent la dégradation des composants de la matrice extracellulaire, limitant ainsi la fibrose. Au niveau hépatique, il est également établi que la sénescence cellulaire accélère la résolution de la situation de cirrhose (fibrose excessive menant à une insuffisance hépatique) induite par les infections virales (hépatites), la consommation excessive d’alcool, ou l’excès de lipides (p. ex. diabète, obésité). Dans l’optique de tirer profits des effets bénéfiques de la sénescence dans le contexte de fibrose hépatique et cardiaque, des protéines CCN1 de synthèse ont d’ores et déjà été testées sur modèles murins, livrant des résultats très prometteurs pour l’utilisation de traitements sénomorphes (induisant la sénescence) chez les patients atteints de cirrhose ou d’infarctus de myocarde.
Implications physiopathologiques et perspectives thérapeutiques
Sénescence et vieillissement
Le vieillissement physiologique se manifeste par une perte progressive de l’homéostasie tissulaire pouvant impacter la physiologie des organes et conduire à l’émergence de pathologies chroniques et liées à l’âge. De nombreuses études rapportent une augmentation du nombre de cellules sénescentes avec l’âge dans divers tissus et organes chez les rongeurs et les primates, dont l’être humain, suggérant un lien de causalité entre sénescence cellulaire et pathologies liées à l’âge.
Historiquement, ce lien fut pour la première fois soulevé à l’issue d’une étude in vivo publiée en 2004, et focalisée sur l’étude d’un modèle murin progéroïde (c'est-à-dire qui présente un vieillissement accéléré) sous-exprimant la kinase BUB1B (mitotic checkpoint serine/threonine kinase B), normalement impliquée dans le contrôle de la ségrégation chromosomique [15]. Ces souris développaient de manière extrêmement précoce de multiples troubles liés à l’âge tels que la cachexie (amaigrissement important), la sarcopénie (diminution des capacités musculaires), la cataracte (opacification du cristallin), la gliose cérébrale (prolifération des cellules gliales) ou encore la perte d’élasticité des parois artérielles. Au travers de manipulations génétiques visant à induire l’apoptose spécifique des cellules sénescentes chez ces souris (modèle murin INK/ATTAC), l’équipe de Jan van Deursen mettait alors en évidence un retard important dans l’émergence de ces divers troubles, sans pour autant que la durée de vie de ces souris ne soit allongée. Quelques années plus tard, en éliminant spécifiquement les cellules sénescentes sur un modèle INK/ATTAC non-progéroïde, cette même équipe notait un retard dans l’émergence des pathologies liées à l’âge accompagné d’une augmentation significative de la durée de vie médiane et maximale, soulignant cette fois-ci l’impact de la sénescence sur l’espérance de vie [16].
Depuis lors, les efforts de recherche sur la sénescence cellulaire se sont orientés sur la dissection des mécanismes cellulaires reliant cette destinée cellulaire et l’émergence des troubles liés à l’âge dans un contexte pathologie-spécifique. Prenons par exemple le cas de l’athérosclérose, cette maladie caractérisée par une perte d’élasticité des artères, avec formation de plaques d’athéromes, des dépôts lipidiques. On sait aujourd’hui que la sénescence joue un rôle ambivalent dans cette pathologie, induisant d’une part l’arrêt de prolifération des monocytes et macrophages infiltrants, réduisant de fait la croissance des athéromes, mais d’autre part favorisant leur rupture via le phénotype sécrétoire associé à la sénescence, avec notamment la libération de métalloprotéases matricielles. Elle est également responsable de la perte d’homéostasie tissulaire observée au niveau des articulations chez les sujets atteints d’ostéoarthrite, de la perte de densité osseuse responsable de l’ostéoporose, de la neuro-inflammation sous-jacente à certaines pathologies neurodégénératives (p.ex. maladie d’Alzheimer et de Parkinson), et contribue au développement du diabète de type 2, de l’obésité, et de certaines formes de fibrose pulmonaire idiopathique (dont la cause est inconnue). Dans ces divers contextes physiopathologiques, le recours à des molécules sénolytiques (p. ex. ruxolitinib, ganciclovir, UBX0101) a d’ores et déjà apporté des résultats particulièrement encourageants sur modèles murins et de nombreux essais cliniques sont en cours chez l’Homme.
Sénescence et cancer : un paradoxe en pleine évolution
Il y a une dizaine d’années, l’hypothèse selon laquelle la sénescence, comme l’apoptose, était un mécanisme suppresseur de tumeur pertinent in vivo manquait d’arguments car elle se fondait uniquement sur les observations faites in vitro. Aujourd’hui, de par l’accumulation de travaux faisant appel à des modèles murins in vivo knock-in et knock-out et de travaux chez l’Homme, le rôle de suppresseur de tumeur que joue la sénescence in vivo est bien confirmé. Aussi, il est établi que le stress engendré par l’hyperactivité d’un oncogène ou la perte d’un gène suppresseur de tumeur engendre, en général, la formation de lésions bénignes constituées de cellules sénescentes. Par ailleurs, des tumeurs malignes, formées de cellules ayant inactivé ou court-circuité le programme de sénescence, se développent à partir de lésions bénignes. Il a notamment été mis en évidence que des souris présentant des défauts majeurs d’apoptose ne sont pas prédisposées de façon marquée à la néoplasie car les cellules protumorales peuvent être éliminées par d’autres mécanismes, dont la sénescence. Cependant, l’inverse n’est pas vrai : des perturbations, même subtiles, des mécanismes de sénescence influencent considérablement la sensibilité au cancer. Ainsi, les souris chez lesquelles un seul allèle des gènes codant p53 ou p16INK4a est altéré sont sujettes aux tumeurs, tandis que les souris portant une copie supplémentaire d’un de ces deux gènes sont résistantes au cancer. Les analyses épidémiologiques pangénomiques les plus récentes ont par ailleurs démontré que la perte de fonction des gènes codant p53 ou p16INK4a est l’événement génétique le plus courant dans les cancers humains, suggérant de fait que le contournement des processus d’entrée en sénescence constitue une étape déterminante de la carcinogenèse. Aussi, des efforts majeurs furent déployés dans la recherche thérapeutique contre le cancer pour rétablir les fonctions de la protéine p53 lorsque le gène associé est altéré – notamment encouragés par les observations stupéfiantes effectuées sur des modèles murins p53 knock-out qui développaient des tumeurs massives régressant rapidement après réactivation de ce gène.
Bien qu’encourageants, les résultats de ces études pionnières publiées dans les années 2000 doivent être nuancés au regard des découvertes les plus récentes. Comme nous avons pu le voir par ailleurs dans d’autres contextes physiopathologiques, la sénescence cellulaire est une destinée cellulaire à deux visages, et dans le contexte particulier du cancer, les études pointant du doigt son potentiel protumorigène s’accumulent. Alors que l’induction de la sénescence des cellules précancéreuses constitue une barrière à la carcinogenèse, ces dernières ont le potentiel de s’accumuler au site de la lésion et de profondément modifier le microenvironnement tumoral via le phénotype sécrétoire associé à la sénescence. Ces molécules sécrétées initient d’une part l’entrée en sénescence des cellules voisines en paracrine via les interleukines IL1α, IL6 ou IL8, mais stimulent d’autre part la progression tumorale en offrant un environnement pro-mitogène aux cellules précancéreuses voisines, et peuvent même induire la transformation des cellules saines et la formation de métastases via le TGFβ et les métalloprotéases matricielles.
À l’heure actuelle, de nombreuses molécules chimiothérapeutiques induisant la sénescence des cellules tumorales sont utilisées en clinique pour lutter contre le cancer (p. ex. docetaxel, bléomycine, cyclophosphamide, doxorubicine, vincristine, étoposide ou encore cisplatine). Néanmoins, l’accumulation des cellules sénescentes qui en résulte au niveau tumoral est souvent associée à une rechute à court ou moyen terme. Aussi, des efforts considérables de recherche sont aujourd’hui dédiés au développement de thérapies en deux étapes (one-two punch therapies), visant à induire la sénescence cellulaire dans un premier temps pour stopper le développement tumoral via des molécules sénomorphes, puis à éliminer rapidement les cellules ainsi devenues sénescentes pour éviter rechute et métastase via des molécules sénolytiques. Certains de ces protocoles one-two punch ont déjà fait preuve de leur efficacité in vivo, comme l’attestent notamment les travaux publiés en 2010 par Clemens Schmidt et son équipe et focalisé sur un modèle murin de lymphome [17]. À l’heure actuelle, se mettent en place de nombreux essais cliniques très prometteurs basés sur ces thérapies combinatoires dans le cadre de la prise en charge des cancers chez l’être humain, redonnant de l’espoir dans la lutte contre ces pathologies dévastatrices.
Glossaire
ATM La protéine ATM (en anglais ataxia telangiectasia mutated) est la protéine mutée dans le syndrome d’ataxie télangiectasie. Elle répare les cassures double-brins dans l’ADN occasionnées par des stress environnementaux ou des processus physiologiques.
ATR La protéine ATR (en anglais ataxia telangiectasia related) est une kinase qui se lie à l’ADN, et inhibe la Cdk1-CyclineB par l’intermédiaire de Chk1 lors du cycle cellulaire.
Autocrine La communication autocrine est un mode de signalisation cellulaire impliquant des messagers chimiques — hormones, cytokines — qui agissent sur la cellule même qui les a synthétisés à travers des récepteurs de la membrane cellulaire.
BRCA1 Le gène BRCA1 (abréviation de breast cancer 1) est un gène humain découvert en 1990 par Mary-Claire King, appartenant à une classe de gènes suppresseurs de tumeur, qui maintiennent l’intégrité génomique afin de prévenir la prolifération incontrôlée de cellules mammaires.
CDC25 Les phosphatases CDC25 à double spécificité (phosphotyrosine et phosphosérine/thréonine) activent les kinases régulatrices du cycle cellulaire (les CDK) et contrôlent, régulabnt ainsi le déclenchement de l’entrée en mitose et la prolifération.
CEBPβ La CCAAT/enhancer-binding protein beta est un facteur de transcription de la famille bZIP jouant un rôle clé dans la régulation de l’expression des gènes de l’inflammation.
DDR Réponse aux dommages à l’ADN.
DSB Cassure double brin de l’ADN.
E2F1 L’E2F1 est une protéine de la famille des E2Fs et jouant le rôle de facteur de transcription. Comme les autres E2Fs, elle intervient dans le cycle cellulaire et a une action de suppresseur de tumeurs.
EGF Le facteur de croissance épidermique est une hormone protéique aux multiples actions, principalement trophiques. La fixation de cette hormone sur le récepteur à l’EGF provoque une activité mitotique très rapide au sein des tissus ciblés.
H2AX H2AFX (famille des histones H2A, membre X) est l’un des plusieurs gènes codant l’histone H2A. H2AX est phosphorylée au niveau de la sérine 139, appelé alors gamma-H2AX, en réaction aux coupures double-brin de l’ADN.
Hétérochromatine L’hétérochromatine est une structure observable de l’ADN, une condensation de la chromatine que l’on distingue de l’euchromatine qui est la structure observable non condensée. L’observation simple de la condensation de la chromatine peut se faire par des colorations de l’ADN notamment avec du DAPI.
HGF Le facteur de croissance des hépatocytes est un facteur de croissance, agent mitogène des hépatocytes (cellules du foie).
Homéostasie En biologie, l’homéostasie est un phénomène par lequel un facteur clé (par exemple, la température) est maintenu autour d’une valeur bénéfique pour le système considéré, grâce à un processus de régulation.
IL1α L’interleukine 1 alpha, aussi connue sous le nom d’hématopoïétine 1, est une cytokine de la famille des interleukine 1. Elle est produite principalement par les macrophages activés, ainsi que par les neutrophiles, les cellules épithéliales et les cellules endothéliales.
IL1β L’interleukine 1 bêta est une isoforme de l’interleukine 1. Elle a un rôle pro- inflammatoire.
IL6 L’interleukine 6 est une cytokine impliquée avec l’IL1β et le facteur de nécrose tumorale (TNF) dans la phase aiguë de l’inflammation. Il s’agit d’une cytokine pro-inflammatoire.
IL8 L’interleukine 8, symbolisée par CX-CL8 ou IL8, est une cytokine, chef de file des chimiokines. Cette molécule est produite en particulier par les cellules épithéliales à la suite de la détection d’agents microbiologiques ou chimiques potentiellement pathogènes. Son rôle principal est d’assurer le recrutement des polynucléaires neutrophiles sur le site de l’infection.
INFɣ Les interférons sont des glycoprotéines de la famille des cytokines. Ils sont naturellement produits par les cellules du système immunitaire, mais également par d’autres types cellulaires (cellules dendritiques, épithéliales …).
Mégacaryocytes Il s’agit d’une cellule géante (d’environ 50 à 100 µm de diamètre) de la moelle hématopoïétique responsable de la production des plaquettes sanguines (ou thrombocytes) lorsque son cytoplasme se fragmente en milliers de plaquettes sanguines (thrombopoïèse, en 4 à 5 jours).
Mésonéphros C’est un organe excréteur rudimentaire qui se développe chez les vertébrés. Chez les mammifères, les oiseaux et les reptiles il représente le deuxième des trois appareils rénaux différents qui se succèdent pendant la vie utérine.
MMP1/3 Les métalloprotéases matricielles sont des protéases, enzymes protéolytiques caractérisées par la présence d’un ion Zn2+ lié à 3 résidus histidine, au niveau de leur site catalytique. Elles ont des rôles dans la modification de la matrice extracellulaire
NFκB Le nuclear factor-kappa B est une protéine de la superfamille des facteurs de transcription impliquée dans la réponse immunitaire et la réponse au stress cellulaire.
OIS Sénescence induite par oncogènes.
p16INK4a Il s’agit d’une protéine suppresseur tumeur, inhibiteur du complexe CDK4/CDK6-Cycline D. Ce complexe phosphoryle pRB ce qui a pour conséquence de l’empêcher de se lier au facteur de transcription E2F. Celui-ci peut alors activer la transcription de nombreux gènes favorisant le passage de la cellule de la phase G1 à la phase S.
p21 Également connue sous le nom de cyclin-dependent kinase inhibitor 1 ou CDK-interacting protein 1, il s’agit d’une protéine inhibitrice des CDK. Elle est connue pour inhiber les complexes CDK2 et CDK4 et permettre ainsi l’arrêt du cycle cellulaire. La protéine p53 est notamment connu pour être un régulateur de p21.
p53 Facteur de transcription régulant de multiples fonctions cellulaires importantes comme le cycle cellulaire, l’autophagie, l’apoptose ou la sénescence. Le gène TP53 codant pour cette protéine est inactivé dans près de 50% des cancers humains.
Paracrine La communication paracrine est un mode de signalisation cellulaire impliquant des messagers chimiques qui agissent dans le voisinage de la cellule qui les a synthétisés.
pRB La protéine du rétinoblastome est une protéine de séquestration qui exerce un contrôle négatif du cycle cellulaire. Cette fonction est essentielle dans les organismes pluricellulaires pour éviter la formation de tumeurs malignes qui mettraient en péril l’organisme, ce qui permet de qualifier cette protéine de suppresseur de tumeur.
ROS Espèces réactive de l’oxygène.
RS Sénescence réplicative.
SAHF Foyers d’hétérochromatine associés à la sénescence.
SASP Phénotype sécrétoire associé à la sénescence.
Sénolytique Qui détruit les cellules sénescentes.
Sénomorphe Qui induit la sénescence cellulaire.
SIPS Sénescence précoce induite par le stress.
SSB Cassure simple brin de l’ADN.
Syncytiotrophoblastes Chez certains mammifères dont l’Homme, le syncytiotrophoblaste est un des tissus qui composent le placenta.
VEGF Le facteur de croissance de l’endothélium vasculaire a pour rôle principal de déclencher la formation de nouveaux vaisseaux sanguins (angiogenèse), nécessaire pour accompagner la croissance des tissus et le développement des organes du corps humain.
β-galactosidase La β-galactosidase est une hydrolase dont le rôle est d’hydrolyser des β-galactosides en oses simples. Ses substrats de prédilection peuvent être le ganglioside GM1, les lactosylcéramides, le lactose, ainsi que plusieurs glycoprotéines. Il s’agit d’un hétérotétramère constitué de quatre sous-unités semblables deux à deux.
Références
- Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
- Lujambio, A. To clear, or not to clear (senescent cells)? That is the question. BioEssays 38, S56–S64 (2016).
- Herranz, N. & Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Invest. 128, 1238–1246 (2018).
- Finch, C. E. Longevity, senescence, and the genome. (University of Chicago Press, 1994).
- Campisi, J. & d’Adda di Fagagna, F. Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740 (2007).
- Sulli, G., Di Micco, R. & di Fagagna, F. d’Adda. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 12, 709–720 (2012).
- Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602 (1997).
- Shay, J. W., Pereira-Smith, O. M. & Wright, W. E. A role for both RB and p53 in the regulation of human cellular senescence. Exp. Cell Res. 196, 33–39 (1991).
- van Deursen, J. M. The role of senescent cells in ageing. Nature 509, 439–446 (2014).
- Muñoz-Espín, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
- Storer, M. et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 155, 1119–1130 (2013).
- Muñoz-Espín, D. et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 155, 1104–1118 (2013).
- Demaria, M. et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
- Yun, M. H., Davaapil, H. & Brockes, J. P. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 4, (2015).
- Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
- Baker, D. J. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
- Reimann, M. et al. Tumor Stroma-Derived TGF-β Limits Myc-Driven Lymphomagenesis via Suv39h1-Dependent Senescence. Cancer Cell 17, 262–272 (2010).