Les téloméropathies forment un spectre de maladies humaines caractérisées par des signes précoces de vieillissement et des extrémités chromosomiques anormalement raccourcies. Ces extrémités, dénommées télomères, ne font pourtant pas partie de l’ADN codant. Comment le rétrécissement des télomères peut-il causer des défaillances d’organes précoces et une prédisposition aux cancers ?
Un des centres d’intérêts intemporels de la biologie est de comprendre les mécanismes intrinsèques du vieillissement. En plus de l’espoir d’augmenter l’espérance de vie humaine, les recherches sur le vieillissement permettent de décoder les origines de nombreuses maladies chroniques dégénératives et de chercher de meilleurs traitements. Le vieillissement se manifeste par une accumulation de déficits structurels et fonctionnels biologiques, physiologiques, et psychologiques d’un organisme vivant (1,2). Il est à la fois irréversible et universel.
Ce déclin de nos fonctions physiologiques est un processus multifactoriel (1). Il peut être résumé par un cumul au cours du temps de dommages cellulaires à plusieurs échelles (entre autres génétiques, épigénétiques, métaboliques et protéiques), réduisant les capacités de réparation tissulaire. De surcroît, des cellules sénescentes, qui habituellement favorisent la réparation des tissus endommagés, s’accumulent avec l’âge et exercent alors un effet inverse en contribuant à diverses dysfonctions tissulaires (cf. partie La sénescence) (1,3,4).
Alors que le terme de « sénescence » provient du latin senescere signifiant « vieillir », le phénomène de sénescence en biologie ne correspond pas véritablement à un vieillissement cellulaire. En vérité, les cellules sénescentes sont des cellules prolifératives ayant arrêté définitivement leur cycle cellulaire sous l’influence de divers signaux de stress (1,3–5). La sénescence cellulaire est ainsi non seulement un mécanisme antiprolifératif permettant de prévenir un développement tumoral suite à des dommages cellulaires, mais aussi un processus crucial dans le remodelage tissulaire, grâce à la sécrétion de facteurs pro-inflammatoires.
Le concept de sénescence cellulaire a été formulé suite à l’observation que des cellules humaines en culture ne pouvaient proliférer à l’infini. En effet, après un certain nombre de mitoses, l’érosion des télomères provoque un arrêt prolongé du cycle cellulaire (1,3–5). Les télomères correspondent aux extrémités des chromosomes. Ils possèdent une structure unique afin de protéger et dissimuler ces terminaisons d’ADN (6–8). À chaque division cellulaire les extrémités chromosomiques sont légèrement raccourcies (cf. partie Les télomères). Lorsque les télomères atteignent une certaine longueur minimale limite, des signaux de stress sont libérés et les cellules entrent en sénescence dénommée réplicative (3–5).
Cependant, certaines cellules de l’organisme sont capables de maintenir la longueur de leurs télomères. C’est le cas des cellules souches, qui assurent le renouvellement continu d’une variété de tissus tout au long de notre vie, comme la peau, l’épithélium intestinal, ou encore le tissu sanguin (3,9,10). Pour prévenir une entrée en sénescence réplicative, et donc une dégradation des tissus qu’elles forment, ces cellules souches expriment l’enzyme télomérase, permettant d’entretenir la longueur de leurs télomères (8,11). Malgré cela, avec l’âge les cellules souches accumulent non seulement des dommages à l’ADN, mais sont aussi victimes d’un raccourcissement progressif de leurs télomères. Ainsi, le nombre et la capacité d’autorenouvellement des cellules souches se dégradent avec l’âge, entraînant le déclin du potentiel régénératif des tissus de l’organisme et contribuant ainsi à son vieillissement (1).
Chez l’Homme, il existe des syndromes de vieillissement prématuré, pour lesquels les individus possèdent des télomères anormalement courts. Ces patients présentent diverses mutations dans les gènes codant les protéines de maintenance des télomères, créant une grande diversité de maladies regroupées sous le nom de téloméropathies (7). Les personnes atteintes de téloméropathies manifestent notamment un déficit précoce de régénération tissulaire, avec une aplasie médullaire (due à une insuffisance de la moelle osseuse), des anomalies de pigmentation de la peau, des cheveux gris précoces voire une alopécie (perte totale des cheveux), ou encore la fibrose de certains organes (poumons, foie) (11,12). L’existence de telles maladies démontre la relation étroite entre vieillissement, sénescence, télomères, cellules souches et réparation tissulaire.
Cet article cherche à tisser le lien entre ces différents phénomènes afin de mieux exposer les causes des téloméropathies, et en quoi ces maladies nous offrent des éclairages sur les origines du vieillissement. Après avoir défini la sénescence cellulaire et son implication dans les processus physiologiques et pathophysiologiques d’un organisme, nous décrirons plus en détail la structure des télomères et des protéines de maintenance des télomères. Cela nous permettra d’établir les fondations pour mieux comprendre les aspects cliniques des téloméropathies, et de faire le tour des dernières connaissances scientifiques sur ce spectre de maladies.
La sénescence
En 1881, le biologiste et médecin allemand August Weismann émit l’hypothèse que les êtres vivants meurent car le renouvellement des tissus biologiques ne peut être infini (8). C’est presque un siècle plus tard que Leonard Hayflick observa que les fibroblastes1 humains en culture ne peuvent proliférer indéfiniment et entrent en sénescence après un nombre défini de divisions cellulaires, connu sous le nom de limite de Hayflick (3,13).

Dommages à l’ADN, développement et érosion des télomères sont autant de mécanismes pouvant conduire une cellule à entrer en sénescence. Même en arrêt de cycle, la cellule reste métaboliquement très active et sécrète des protéines du phénotype sécrétoire associé à la sénescence (SASP), pouvant induire une entrée en sénescence de cellules voisines (sénescence paracrine), du remodelage tissulaire, et le recrutement de cellules immunitaires. Schéma réalisé avec BioRender.com.
La sénescence est une reprogrammation cellulaire caractérisée par un arrêt du cycle cellulaire et une importante activité métabolique. Les cellules sénescentes sécrètent une variété de protéines constitutives du phénotype sécrétoire associé à la sénescence (Senescence Associated Secretory Phenotype, ou SASP), qui induisent, suivant le contexte, un remodelage tissulaire avec notamment la formation de nouveau vaisseaux sanguins (angiogénèse), le recrutement de cellules immunitaires, ou encore l’entrée en sénescence des cellules avoisinantes (3) (Figure 1).
Contrairement à ce qu’on pourrait penser, les cellules sénescentes ne sont pas seulement liées à des pathologies ou au vieillissement de notre organisme. En effet, le phénomène de sénescence apparaît dès le développement embryonnaire, par exemple en contribuant à la morphogénèse de l’oreille interne (14). Puis, après la naissance, la sénescence est induite physiologiquement en réponse à des dommages causés à l’ADN, par exemple dans le cadre d’une plaie, où il a été démontré que la présence de cellules sénescentes favorise la cicatrisation par la sécrétion du platelet-derived growth factor AA (PDGF-AA) facilitant le remodelage tissulaire et l’angiogénèse (15). Ainsi, l’accumulation de dommages à l’ADN et de stress cellulaires peuvent conduire une cellule à entrer en sénescence, plutôt qu’à subir une transformation cellulaire pouvant amener à un développement tumoral. La sénescence des cellules est ainsi considérée comme un mécanisme de protection contre le cancer, d’autant plus que les cellules sénescentes sécrètent des cytokines pro-inflammatoires attirant des cellules immunitaires. Ces dernières éliminent à la fois les cellules sénescentes, et le tissu environnant ayant le potentiel de devenir précancéreux après une exposition à divers stress cellulaires (16).
À l’opposé, les cellules sénescentes ont aussi des effets néfastes sur l’organisme, notamment en s’accumulant avec l’âge. L’élimination des cellules sénescentes, via des traitements médicamenteux ou des modèles murins transgéniques, retarde l’apparition de dysfonctions tissulaires liées à l’âge telles que l’athérosclérose1 ou la fibrose pulmonaire2 (3,17). Par ailleurs, certaines molécules du SASP sont des cytokines pro-inflammatoires, qui sont suspectées de participer à l’inflammation liée à l’âge, aussi dénommée inflammaging. Ce déclin est exacerbé par l’entrée en sénescence des cellules immunitaires elles-mêmes et par la diminution de la quantité de cellules souches hématopoïétiques, qui réduisent l’efficacité du système immunitaire et donc l’élimination des cellules sénescentes de l’organisme (3). Ce phénomène est aussi connu sous le nom d’immunosénescence. La réduction du nombre de cellules souches hématopoïétiques et neurales a été corrélée avec une augmentation de certains marqueurs de sénescence dans les régions où résident ces cellules, il est donc envisageable que la sénescence participe aussi à la diminution du renouvellement de certains tissus de l’organisme (1,3).
Ainsi, même si les cellules sénescentes jouent un rôle clé dans de nombreux phénomènes physiologiques, elles peuvent aussi être néfastes dans certains contextes pathologiques. L’accumulation de cellules sénescentes avec l’âge participe à diverses dysfonctions tissulaires, contribuant au vieillissement irréversible de l’organisme. L’érosion des télomères est un des mécanismes induisant la sénescence cellulaire, et ce phénomène entretient aussi un lien étroit avec le processus de vieillissement.
Les télomères
Lorsqu’une cassure double brin se produit dans le génome, elle est réparée par des mécanismes spécifiques afin de relier les extrémités séparées. Or, le génome humain est divisé en 46 chromosomes linéaires, chacun d’entre eux possédant deux extrémités qui ressemblent justement à des cassures double brin. Pourtant, comme le mit en évidence Hermann Muller en 1938, ces extrémités ne sont pas sujettes aux mécanismes de réparation des cassures double brin (8). Il nomma ces extrémités télomères, du grec telos signifiant « fin » et meros signifiant « partie ». À la même époque, Barbara McClintock observa que les extrémités chromosomiques des cellules de maïs étaient sous forme d’hétérochromatine, autrement dit que leur ADN était extrêmement condensé, suggérant une activité transcriptionnelle réduite (8).
Structure des télomères
Les télomères correspondent à une structure singulière de l’ADN aux extrémités des chromosomes. Ils sont constitués de répétitions en tandem3 (TTAGGG)n chez les mammifères, et d’un ensemble protéique protégeant cet ADN, formant le complexe dénommé shelterine (de l'anglais shelter, abri). Si les extrémités chromosomiques ne sont pas reconnues comme des cassures double brin par les machineries de réparation de l’ADN, c’est parce que les télomères sont dissimulés par divers mécanismes de protection, dont l’un des plus étudiés est une structure en boucle appelée t-loop (8,11).
Le complexe shelterine est constitué de différentes protéines (Figure 2). Les telomere repeat binding factor 1 et 2 (TRF1 & TRF2) se fixent sur les répétitions (TTAGGG)n double brin et interagissent avec le repressor/activator protein 1 (RAP1). La protéine protection of telomeres 1 (POT1) se fixe quant à elle sur l’ADN simple brin et interagit indirectement avec TRF1 et TRF2 via TRF1-interacting nuclear protein 2 (TIN2) et TIN2-interacting protein 1 (TPP1). Enfin, le simple brin dépassant à l’extrémité du télomère effectue une boucle et s’hybride avec le brin complémentaire sur une zone située en amont, ce qui le rend inaccessible aux machineries de réparation de l’ADN (8,11).
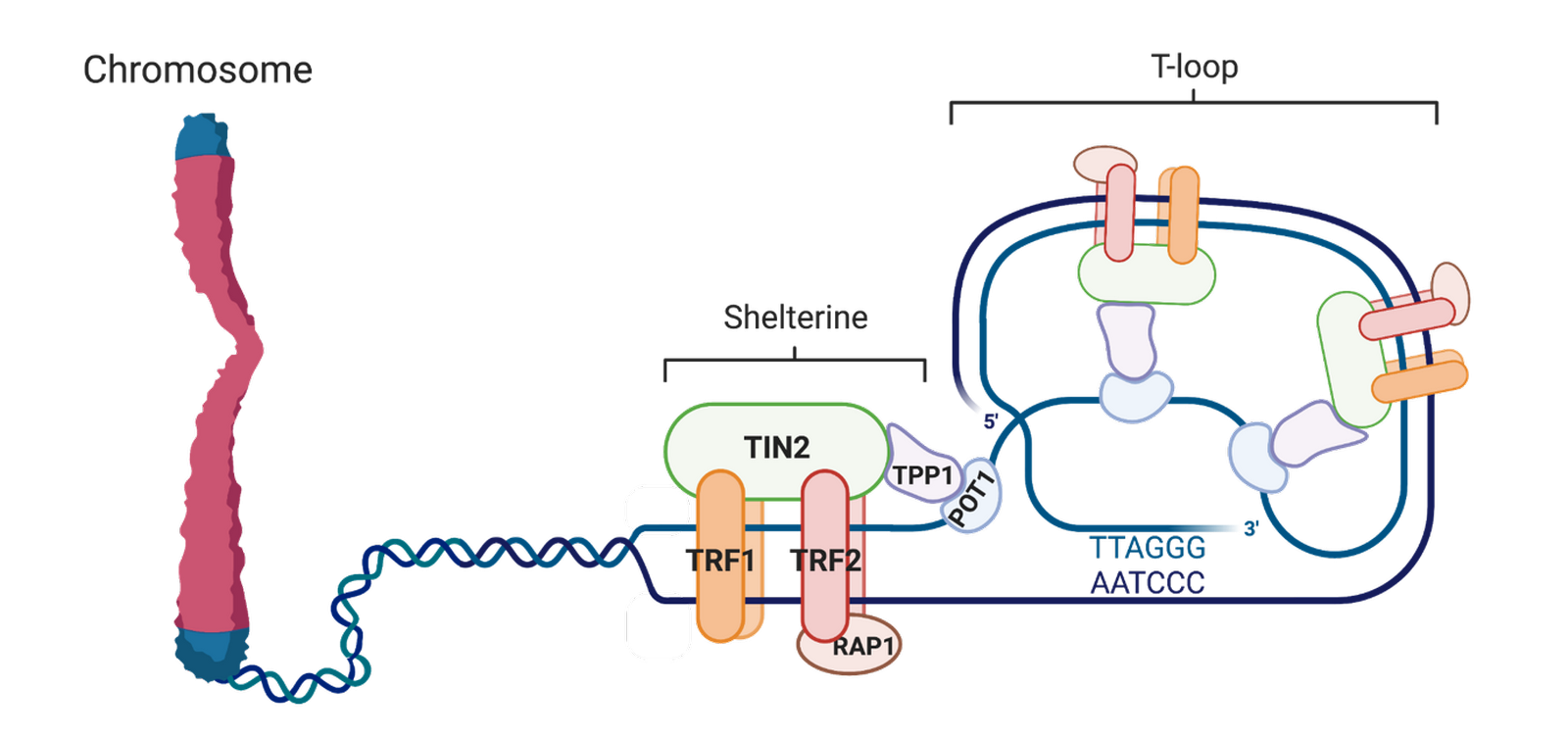
Représentation schématique d’un télomère humain. Le complexe protéique shelterine réarrange l’extrémité chromosomique en une t-loop qui dissimule la terminaison libre du chromosome. Schéma réalisé avec BioRender.com.
Le simple brin d’ADN présent à l’extrémité des télomères provient de la réplication asymétrique du matériel génétique. Lors de la duplication de l’ADN au cours du cycle cellulaire, un des brins est copié dans le sens inverse de celui de l’avancée de la fourche de réplication. Ce brin est donc synthétisé de manière discontinue, par segments dénommés fragments d’Okasaki (13). Par conséquent, à l’extrémité du brin discontinu, une amorce ARN ne peut se fixer en 5’ pour terminer la réplication du brin, et à chaque division cellulaire les télomères se raccourcissent progressivement (Figure 3). En parallèle, le brin continu subit un raccourcissement de son extrémité 5’ après avoir été synthétisé. En effet, la protéine POT1 doit se fixer sur un simple brin d’ADN pour que la structure t-loop puisse se former et protéger le télomère. Or, la synthèse du brin continu se déroule jusqu’à l’extrémité 5’ du brin parent, ne laissant aucun segment en simple brin. La nucléase Apollo résèque donc l’extrémité 5’ du brin parent afin de former un surplomb télomérique (« G-overhang ») (18), ce qui contribue aussi au raccourcissement progressif des télomères. Lorsque les télomères atteignent une certaine longueur minimale critique, les cellules entrent en sénescence réplicative, à l’origine de la limite de Hayflick (13).
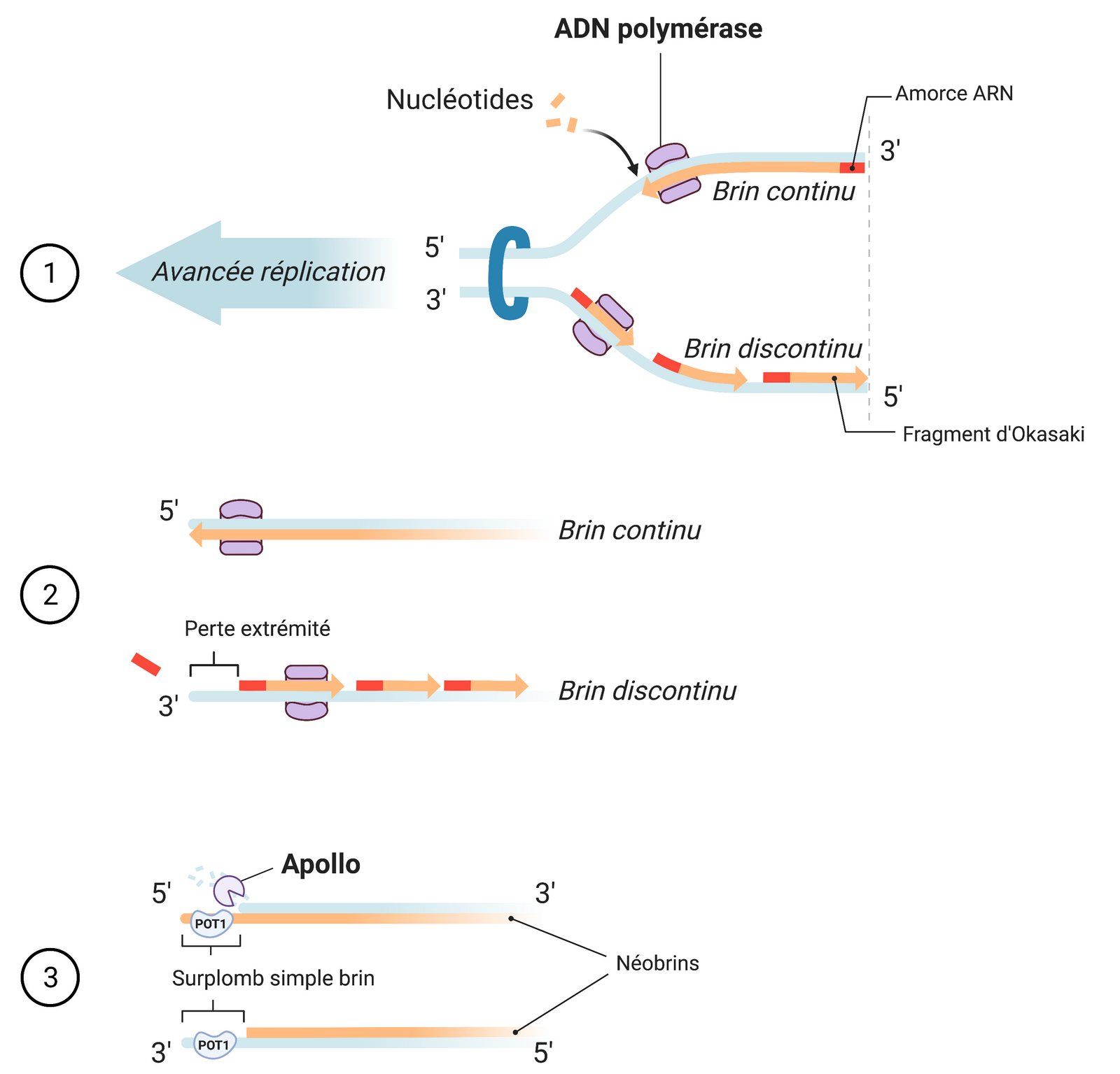
Les deux brins d’ADN sont séparés par une topoisomérase, et deux néobrins complémentaires sont formés à partir des brins parentaux par l’ADN polymérase. Le brin direct est synthétisé de manière continue, tandis que le brin indirect doit être synthétisé de manière discontinue, par fragments d’Okasaki. Les nouvelles extrémités gardent un surplomb simple brin où la protéine POT1 peut se fixer pour permettre la formation de la structure t-loop. Schéma réalisé avec BioRender.com.
L’enzyme télomérase
Même si la majorité des cellules somatiques de l’organisme à l’âge adulte sont différenciées et restent en phase G0 de leur cycle cellulaire (19), certains tissus conservent des cellules souches proliférant tout au long de la vie. La peau se renouvelle chaque mois (9), l’épithélium intestinal tous les deux à trois jours (10), les hématies tous les quatre mois (16), sans oublier la spermatogenèse qui dure environ deux mois (20), pour ne citer que quelques exemples. S’il n’y avait aucun mécanisme empêchant le raccourcissement des télomères, les cellules souches de l’organisme entreraient rapidement en sénescence, ce qui entraînerait de nombreuses dysfonctions tissulaires.
Cependant, les tissus germinaux ont la particularité de posséder des télomères plus longs que les tissus somatiques (8), grâce à l’activité de l’enzyme télomérase permettant de maintenir la longueur des télomères. Elle est exprimée physiologiquement dans les cellules souches du corps humain et dans 85-90 % des cancers, autrement dit dans les cellules hautement prolifératives. Les cellules cancéreuses présentent en revanche des télomères plus courts que les cellules germinales, ce qui contribue à leur instabilité génomique (8,21). Ce phénomène paradoxal s’explique par le fait que la télomérase occupe seulement une minorité des télomères, les plus courts, pendant que d’autres télomères continuent de se raccourcir à chaque division cellulaire, résultant en une diminution de la longueur moyenne de l’ensemble des télomères des cellules cancéreuses (21).
La découverte de la télomérase revient à Carol Greider et Elizabeth Blackburn, qui ont reçu le prix Nobel de physiologie ou médecine en 2009 (8). La télomérase est un complexe protéique composé de l’enzyme telomerase reverse transcriptase (TERT), d’une amorce ARN appelée telomerase RNA component (TERC) servant de matrice pour l’ajout des répétitions (TTAGGG)n, et d’un complexe dyskérine aidant au bon fonctionnement de l’enzyme télomérase et à la stabilisation de l’amorce ARN (8,11) (Figure 4). La télomérase possède une activité de type transcriptase inverse, permettant de rallonger le brin G (celui se terminant par un simple brin en 3’). Puis les ADN polymérases alpha et delta, dont la première présente également une activité primase, se servent du brin G comme matrice pour allonger le brin C (celui avec une extrémité 5’). L’expression de la télomérase est régulée par des facteurs de transcription et par divers mécanismes épigénétiques, afin de limiter son expression à certaines cellules en période postnatale (21).
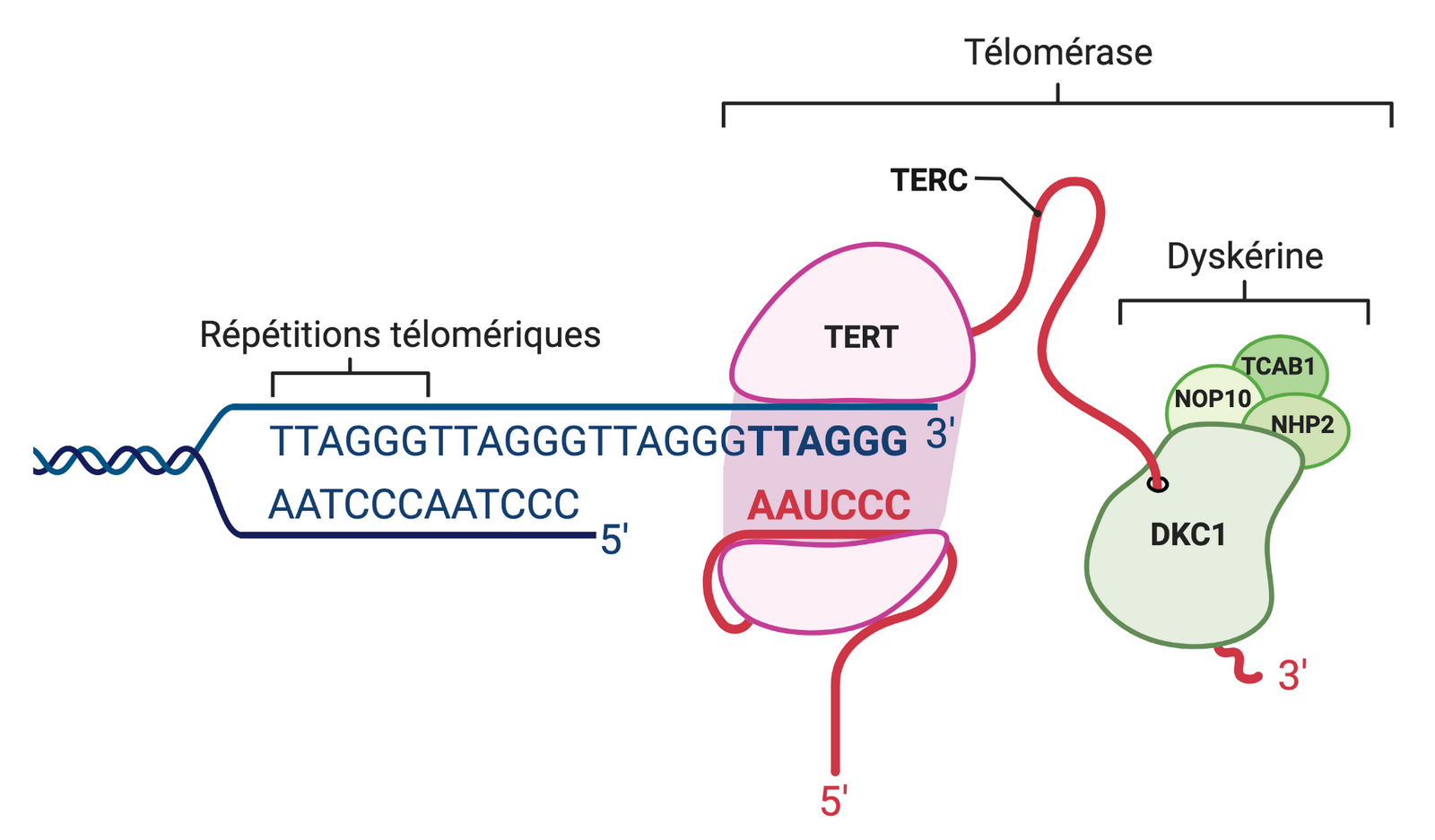
Le complexe télomérase permet de rallonger les télomères dans les cellules eucaryotes. Une amorce ARN, nommée TERC, s’hybride spécifiquement aux répétitions (TTAGGG)n télomériques. Le complexe est composé principalement d’une transcriptase inverse TERT (sous-unité catalytique) et de protéines adjacentes essentielles à la stabilisation du complexe protéique. Schéma réalisé avec BioRender.com.
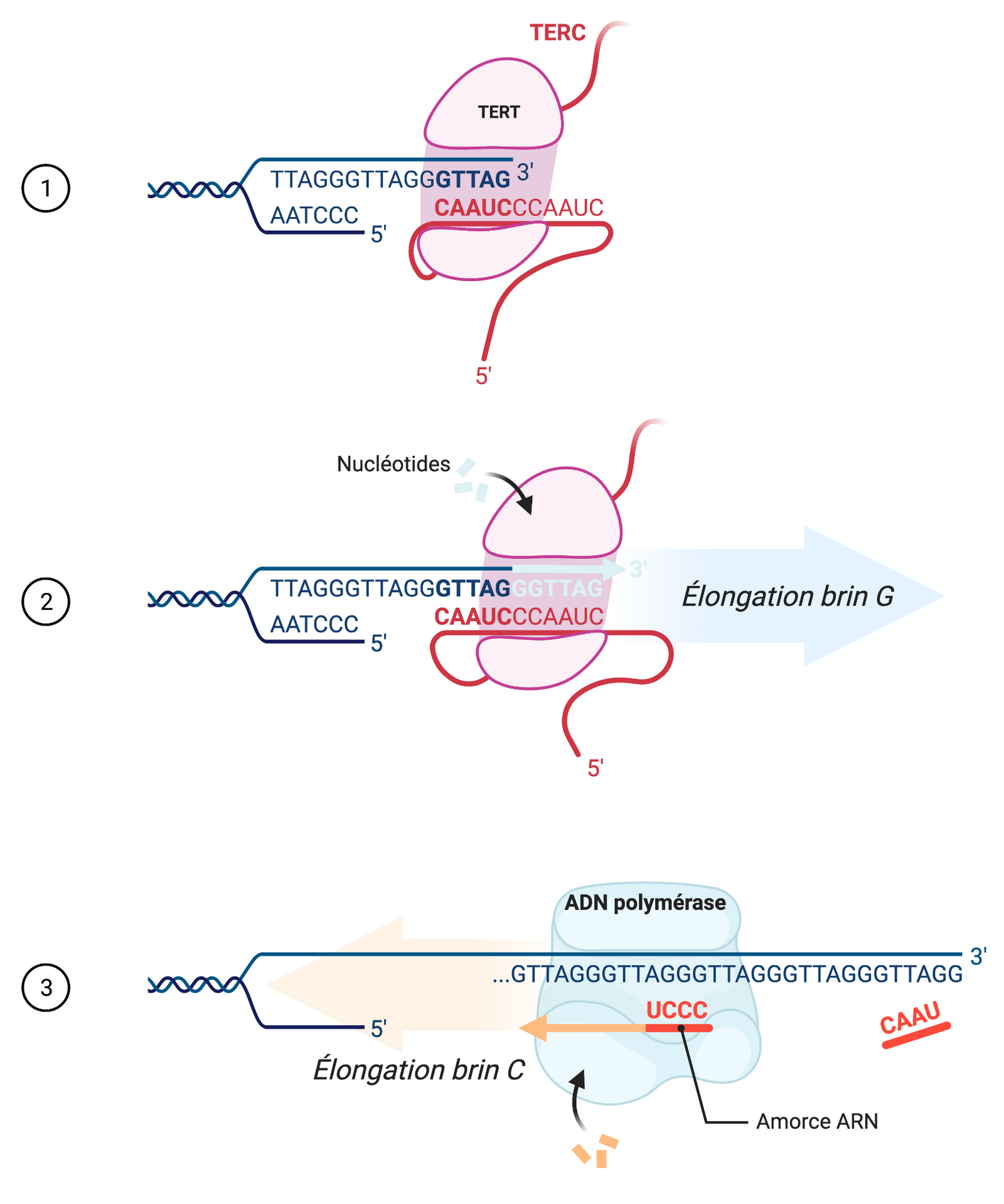
Le complexe télomérase initie le rallongement du télomère. TERC s’hybride en complémentarité de l’extrémité 3’ du brin G (1), puis la transcriptase inverse TERT rallonge le brin (2). Par la suite, les ADN polymérases alpha et delta synthétisent de manière discontinue le brin C complémentaire (3). Schéma réalisé avec BioRender.com.
Ainsi, les télomères permettent non seulement de protéger les extrémités chromosomiques, mais aussi de servir de « zone tampon » pour éviter une perte de matériel génétique au cours des divisions cellulaires. Malgré l’existence de mécanismes de maintenance de la longueur des télomères, avec l’âge les cellules souches perdent progressivement leurs télomères, et il est possible que cela participe à l’apparition de cellules sénescentes parmi la réserve de cellules souches (22). L’érosion des télomères, la sénescence cellulaire et l’épuisement de la réserve de cellules souches sont à ce jour considérés comme trois phénomènes plus ou moins interdépendants, contribuant au vieillissement des tissus biologiques (1).
Au cours des vingt dernières années, les scientifiques ont progressivement associé certains syndromes de vieillissement précoce à un dysfonctionnement de maintenance des télomères. Certains individus présentaient des télomères anormalement courts, et l’avancée des connaissances scientifiques sur la structure des télomères et leurs fonctions a permis de découvrir que ces patients avaient des mutations dans divers gènes liés à la maintenance des télomères (11). Chez ces individus, les anomalies de leurs télomères coïncidaient avec des tableaux cliniques extrêmement variés en termes de phénotype et de sévérité. Les syndromes cliniques dus à des télomères anormalement courts ont ainsi été regroupés dans un spectre de maladies dénommées téloméropathies.
Les téloméropathies
L’importance des télomères et de la télomérase a été mise au jour notamment avec l’identification de maladies héréditaires monogéniques se manifestant par des signes de vieillissement prématuré et un raccourcissement notable des télomères (8,12,23). Les individus portant une mutation dans toutes les cellules de leur corps (mutation constitutionnelle) dans des gènes de maintenance des télomères développent une variété de maladies touchant ainsi différents organes (poumon, moelle osseuse, foie, peau…), et apparaissant à différents âges. C’est pourquoi ces syndromes ont été regroupés sous le nom de spectre de défaut de maintenance des télomères, aussi connu en tant que téloméropathies.
La complexité de présentation des téloméropathies rend leur classification difficile. Un outil d’aide au diagnostic des téloméropathies est la mesure de la taille des télomères par Flow Fluorescent In-Situ Hybridization (flow-FISH), à partir de prélèvements sanguins. Une sonde fluorescente s’hybride spécifiquement aux répétitions télomériques TTAGGG (FISH), puis la fluorescence émise dans le noyau des cellules est quantifiée par cytométrie de flux (flow). La longueur moyenne de l’ensemble des télomères, proportionnelle à l’intensité de la fluorescence, est ensuite exprimée en percentile par rapport à une population de référence du même âge que l’individu. Grâce à l’amélioration des techniques de biologie moléculaire, les téloméropathies sont de mieux en mieux diagnostiquées en France. En 2019, l’incidence était estimée à 0,6 pour 100 000 habitants (24).
Pour ces maladies, on décrit une pénétrance incomplète, c’est-à-dire que les individus portant une mutation sur le même gène n’ont pas le même risque de manifester la maladie à un âge donné. Les téloméropathies ont aussi une expressivité variable, signifiant que les patients ont des présentations cliniques multiples (12). De manière générale, une téloméropathie peut être diagnostiquée à tout âge de la vie, mais une apparition précoce des symptômes est corrélée à une forme plus sévère de la maladie (23,24).
Téloméropathies se manifestant à l’âge adulte
Chez l’adulte, la fibrose pulmonaire idiopathique est la forme la plus fréquente de téloméropathie, apparaissant chez les patients de 50 à 60 ans (12,24,25). Elle est caractérisée par une dégénérescence progressive du poumon due à une inflammation chronique et au développement d’une fibrose. Parmi les formes familiales de cette maladie, un tiers des familles portent une mutation (principalement à l’état hétérozygote) du complexe télomérase, notamment sur l’enzyme TERT ou l’acide ribonucléique TERC (12,23–25). Il est à noter que les mutations de TERC ou TERT sont, dans cette présentation pulmonaire, majoritairement de transmission autosomique dominante avec une haploinsuffisance de la télomérase. En d’autres termes, un des allèles est fonctionnel (allèle sauvage), mais celui-ci n’est pas suffisant pour compenser complètement l’allèle déficient (allèle muté). Cette haploinsuffisance a été proposée pour expliquer l’apparition « tardive » des premiers symptômes dans les premières générations des familles mutées (26).
Étant donné que la fibrose pulmonaire idiopathique est la manifestation la plus fréquente des téloméropathies, les scientifiques ont cherché à mieux comprendre comment la déficience d’une protéine de maintenance des télomères pouvait causer une dégénérescence pulmonaire accélérée. Ainsi, une étude a démontré que la suppression conditionnelle de Trf2, codant une protéine du complexe shelterine (Figure 2), dans les pneumocytes de type II chez des souris adultes induisait la sénescence des cellules souches alvéolaires, ainsi que la sécrétion de cytokines pro-inflammatoires typiques d’une inflammation pulmonaire (27). Les souris présentaient plusieurs dysfonctions pulmonaires, et après exposition à la bléomycine (agent pneumotoxique), aucune n’a survécu plus de 30 jours, tandis que 50 % des souris témoins ont survécu plus de 2 mois (27). Cette étude a ainsi mis en évidence qu’une perturbation télomérique peut induire l’entrée en sénescence de cellules souches pulmonaires, impactant les capacités régénératives du poumon et mettant en danger la survie de l’hôte en présence de substances pneumotoxiques. Pour autant, il reste encore à démontrer si la fibrose pulmonaire idiopathique chez des patients avec une téloméropathie est due ou non à une déficience des cellules souches alvéolaires.
D’autres atteintes dues à un dysfonctionnement télomérique se manifestant à l’âge adulte incluent l’aplasie médullaire1 et la cirrhose cryptogénétique2. L’insuffisance médullaire peut se présenter de différentes façons : un déficit immunitaire, des hématies de diamètre anormalement large (macrocytose), un manque de plaquettes (thrombopénie), qui peuvent évoluer vers des anomalies plus sévères dans plusieurs lignées sanguines (myélodysplasie, leucémie myéloïde) (24,28). Quant aux atteintes hépatiques, leur prévalence est estimée à moins de 10 %, mais elles sont probablement sous-estimées (24). Des résultats récents, entre autres sur des séries de patients avec présentation hépatique, semblent montrer que le spectre des atteintes est large (anomalie du bilan biologique hépatique, maladie vasculaire du foie ou cirrhose), et pas si rare (29). À noter que les patients peuvent développer plusieurs atteintes simultanément (fibrose pulmonaire, cirrhose hépatique et aplasie médullaire) (2,12,24), suggérant que le raccourcissement des télomères se produit dans plusieurs organes, entraînant leur vieillissement prématuré.
Malgré l’apparition « tardive » de ces différentes conditions, dès le diagnostic d’une atteinte pulmonaire, hépatique ou hématologique, la survie du patient portant une mutation des gènes de maintenance des télomères est évaluée à moins de 5 ans (24). C’est pourquoi un dépistage précoce de mutations délétères dans des gènes de maintenance des télomères grâce à un diagnostic génétique d’apparentés, permet d’élaborer un plan de prévention pour limiter l’exposition à des substances toxiques environnementales tels que le tabac et la pollution atmosphérique (12,24), qui accéléreraient l’émergence des défaillances d’organes.
Téloméropathies précoces
Caractéristiques phénotypiques
La première maladie identifiée liée à un défaut de maintenance des télomères fut la dyskératose congénitale. Cette maladie est caractérisée par trois signes cliniques principaux : une dysplasie unguéale (morphologie anormale des ongles), une leucomélanodermie réticulée (anomalies de pigmentation de la peau), et une leucoplasie buccale (lésions blanches dans la bouche) (12,23,30). Les symptômes cutanés et unguéaux apparaissent généralement vers l’âge de 10 ans, puis une aplasie médullaire se développe avant l’âge de 30 ans chez 80 à 90 % des patients (31). Lorsqu’un dépistage de téloméropathie est effectué, la plupart de ces patients ont une longueur télomérique inférieure au premier percentile en flow FISH (23,24). D’autres symptômes variés peuvent apparaître : retards de développement, petite taille, fibrose pulmonaire, canitie et/ou alopécie précoces3, caries dentaires, ostéoporose, diminution du diamètre (sténose) de l’œsophage, pour ne citer que les plus fréquents (12,31,32).
En revanche, malgré la pénétrance incomplète de ces différents symptômes, la plupart des patients présenteront des défaillances d’organes présentant des tissus à renouvellement rapide (peau, moelle osseuse, système digestif). D’ailleurs, la mortalité précoce élevée des patients avec une dyskératose congénitale est due à une défaillance d’organes dans plus de 80 % des cas, notamment par des complications médullaires et/ou pulmonaires (31).
De surcroît, environ 10 % des patients développent un cancer entre 30 et 40 ans (11,23,31,32), qui touche le plus souvent les tissus à renouvellement fréquent. Les téloméropathies précoces telles que la dyskératose congénitale ont une incidence accrue de carcinome épidermoïde de la tête et du cou (cancer de la peau), de syndrome myélodysplasique et de leucémie myéloïde aiguë (cancers de la moelle osseuse).
Il peut paraître contre-intuitif que des patients avec des télomères extrêmement courts puissent développer des cancers, étant donné qu’une longueur de télomère réduite limite la prolifération cellulaire, comme nous l’avons vu avec la sénescence réplicative. Malgré cela, l’érosion sévère des télomères suscite des signaux de dommages à l’ADN, et peut amener à des fusions chromosomiques ou réarrangements géniques (11,12). De plus, l’expression des gènes adjacents aux régions télomériques peut être réprimée, par un effet de position télomérique. Cette instabilité génique favorise l’apparition d’une minorité de cellules précancéreuses qui, dans le contexte d’une téloméropathie, auront un avantage prolifératif dans une niche appauvrie en cellules souches (12,23). Il faut ajouter à cela l’immunosénescence précoce de ces patients, causant un déficit de surveillance immunitaire de tissus précancéreux (11,23). Par la suite, afin de conserver leur capacité d’autorenouvellement et leur prolifération accrue, les cellules cancéreuses sont capables d’entretenir la longueur de leurs télomères via divers mécanismes. La grande majorité des cancers réexpriment l’enzyme télomérase (33), et une minorité emploient un mécanisme d’élongation alternative des télomères (Alternative Lenghtening of Telomeres, ALT) se basant sur une recombinaison intertélomérique (34).
Il existe d’autres phénotypes plus sévères de dyskératose congénitale, se manifestant dès la naissance, voire en prénatal. Le syndrome de Høyeraal-Hreidarsson se traduit par un retard de croissance intra-utérin, un déficit immunitaire, une anémie aplasique, une microcéphalie4 et une hypoplasie cérébelleuse5 (24,35,36). Le syndrome de Revesz est caractérisé par des symptômes similaires, avec de plus une rétinopathie6. Enfin, le syndrome de Coats plus peut présenter des symptômes des deux précédents syndromes, en plus de calcifications cérébrales (11,12). Malgré leur sombre pronostic, ces syndromes restent extrêmement rares. Par exemple, la dyskératose congénitale ne touche qu’un individu sur un million (11).
Héritabilité des téloméropathies précoces
La dyskératose congénitale est le plus souvent caractérisée par une diminution de la longueur des télomères. Différentes mutations et modes de transmission ont été découverts, mais il reste nécessaire d’approfondir les recherches sur cette maladie afin de mieux comprendre la grande variabilité des symptômes.
La dyskératose congénitale liée à l’X est due à une mutation du gène DKC1 codant la protéine dyskérine, un des composants de la télomérase (Figure 4). Ce gène étant situé sur le chromosome X, une mutation délétère de ce gène a donc un mode de transmission récessif lié à l’X. Dans les familles portant une mutation du gène DKC1, la dyskératose congénitale se manifeste dans la grande majorité des cas chez les hommes (31,32,37). Étant donné que les femmes ont deux chromosomes X, elles doivent hériter de deux allèles défectueux du gène DKC1, ou bien avoir un biais d’inactivation du chromosome X portant l’allèle sain, pour développer des symptômes de téloméropathie. Environ 20 à 25 % des cas de dyskératose congénitale sont dus à une mutation du gène DKC1 (Tableau 1).
Une autre étiologie fréquente de la dyskératose congénitale est une mutation hétérozygote du gène TINF2, codant la protéine TIN2 faisant partie du complexe shelterine (Figure 2A). Étant donné l’importance cruciale de la shelterine pour maintenir la structure des télomères, des cellules portant un défaut dans un de ses composants auraient une instabilité génétique précoce qui les mettrait sous une forte pression de sélection négative (12). C’est pourquoi les dyskératoses congénitales dues à une mutation TINF2 ont un mode de transmission autosomique dominant, sont très fréquemment de novo (la mutation n’est pas présente chez les parents), et se manifestent dès la première décennie de la vie du patient, soit sous une forme sévère (syndrome de Høyeraal-Hreidarsson ou de Revesz), soit avec une insuffisance médullaire (32,38,39). Environ 15 à 20 % des dyskératoses congénitales sont dues à une mutation TINF2 (Tableau 1).
Par ailleurs, les mutations des gènes TERC ou TERT ont le plus souvent un mode de transmission autosomique dominant, et expliquent environ 10 % des cas de dyskératose congénitale (11,32). Plusieurs études fonctionnelles ont reproduit certaines mutations dans des lignées cellulaires, et ont prouvé que les mutations TERC présentes dans la dyskératose congénitale entraînent une déficience de l’activité de la télomérase soit par une instabilité accrue de l’amorce TERC, soit par un défaut catalytique (40). Une haploinsuffisance de l’enzyme TERT est suffisante pour entrainer un raccourcissement accéléré des télomères, causant une perte de cellules souches, notamment dans les tissus à renouvellement fréquent tels que la moelle osseuse et la peau (12,31). Pour le gène TERT, codant la rétrotranscriptase du complexe, une perte de fonction hétérozygote se manifeste plutôt chez des adultes sous forme de fibrose pulmonaire familiale, à transmission autosomique dominante, tandis que des mutations bialléliques sont plutôt corrélées à une dyskératose congénitale avec transmission autosomique récessive (23,32).
Enfin, une minorité des cas de dyskératose congénitale ont une transmission autosomique récessive.
Un petit nombre de familles ont été décrites dans la littérature ayant une mutation d’autres composants de la télomérase : NHP2, NOP10 et TCAB1 (Figure 4) (32,35,37).
D’autres cas de dyskératose congénitale sont associés à un défaut de réplication des télomères. Lors de la réplication de l’ADN, la structure t-loop des télomères doit être dissociée, sans que les séquences répétitives télomériques puissent former des structures secondaires telles que des G-quadruplex7, qui bloqueraient la fourche de réplication (12). La protéine RTEL1 est une ADN hélicase importante pour maintenir la stabilité des télomères au cours de leur réplication. Des mutations RTEL1 ont été décrites dans le syndrome de Høyeraal-Hreidarsson à un état biallélique et dans la fibrose pulmonaire idiopathique à un état monoallélique (36,41). Par ailleurs, la protéine conserved telomere protection component 1 (CTC1) participe au redémarrage des fourches de réplication télomériques bloquées, et à l’élongation du brin C (celui avec une extrémité 5’) par le complexe télomérase. Une déficience en CTC1 dans un modèle murin a démontré un raccourcissement accéléré du brin C des télomères, ainsi qu’une insuffisance médullaire précoce (42). Environ 90 % des patients avec un syndrome Coats plus ont une mutation biallélique du gène CTC1 (Tableau 1) (11,32).
Malgré les progrès de dépistage et de caractérisation génotypique des cas de dyskératose congénitale durant les 20 dernières années, à ce jour environ 40 % des cas de dyskératose congénitale restent inexpliqués (11,23,32).
| Expression clinique | Gène | Distribution |
|---|---|---|
| Dyskératose congénitale Høyeraal-Hreidarsson |
DKC1 | 20-25 % |
| TINF2 | 15-20 % | |
| TERT, TERC | ~10 % | |
| RTEL1 | 2-8 % | |
| PARN | 1-5 % | |
| NHP2, NOP10, TCAB1, ACD (codant TPP1) | rares | |
| Syndrome de Coats plus | CTC1 | ~90 % |
| POT1, STN1 | rares | |
| Fibrose pulmonaire familiale | TERT, TERC | 10-20 % |
| RTEL1 | ~5 % | |
| PARN | 1-5 % | |
| DKC1, TINF2, NHP2, NOP10, ACD (codant TPP1), NAF1, ZCCHC8 | rares | |
| Aplasie médullaire | TERT, TERC | 3-5 % |
| RTEL1 | ~1 % | |
| DKC1, PARN, TINF2, ACD (codant TPP1) | rares |
La distribution représente la proportion de patients avec une expression clinique donnée, qui possèdent une mutation du gène désigné (ex. : 10 à 20 % des patients avec une fibrose pulmonaire familiale ont une mutation du gène TERT et/ou TERC). Données issues de (8,11,12,32,37,41).
Téloméropathies : un spectre de maladies multifactorielles
De nombreux aspects des téloméropathies restent à ce jour encore inexpliqués, en particulier le lien entre génotype et phénotype. En effet, les phénotypes ont une pénétrance incomplète (24,32,37,41). À titre d’exemple, parmi différentes familles portant une mutation sur le même résidu de la protéine TIN2 (l’arginine 282), la triade classique de la dyskératose congénitale n’apparaît pas systématiquement, et différents syndromes peuvent se présenter, entre aplasie médullaire et syndrome de Høyeraal-Hreidarsson (43).
Par ailleurs, un même phénotype peut être causé par différentes mutations. Par exemple, l’aplasie médullaire se manifeste dans la plupart des téloméropathies, à différents âges, que ce soit dû à des mutations de TERT, TERC, TIN2, ou encore de TCAB1 (28,31,32).
Afin d’expliquer une telle hétérogénéité dans ce spectre de défaut de maintenance des télomères, il faut considérer la prédisposition génétique des patients, la longueur des télomères hérités, et leur exposition à divers facteurs de risques environnementaux.
Prédisposition génétique
Comme nous l’avons vu précédemment, l’origine des téloméropathies, quelles qu’elles soient, reste un défaut dans un gène de maintenance des télomères. Après un certain nombre de divisions successives, les cellules perdent leurs télomères et entrent en sénescence ou en apoptose (11,23). À l’échelle de l’organisme, la perte progressive de la population de cellules souches amène à une succession de dysfonctions tissulaires, causant un vieillissement précoce et une mortalité élevée.
La moelle osseuse en est le meilleur exemple : environ un milliard de cellules sanguines sont produites chaque heure (11). Dans un tissu avec un tel taux de renouvellement, il n’est pas surprenant qu’un défaut de longueur des télomères entraîne tant de dysfonctionnements. Ainsi, le lien de cause à effet entre télomères raccourcis et épuisement de la réserve de cellules souches hématopoïétiques a été clairement établi à la fois chez l’être humain et dans des modèles murins (28,44).
Un défaut de maintenance des télomères peut aussi entraîner des dysfonctions tissulaires sans perte de la population cellulaire. L’instabilité télomérique déséquilibre l’homéostasie cellulaire avec des répercussions sur le métabolisme, via divers signaux de dommages à l’ADN et changements de l’expression génique. Par exemple, dans un modèle murin avec des télomères anormalement courts et une délétion hétérozygote Terc, les souris adultes ont un défaut de sécrétion d’insuline et une intolérance au glucose, malgré une abondance conservée des cellules β pancréatiques8 (45). Ces cellules β manifestaient plusieurs caractéristiques de sénescence, et présentaient ex vivo une dysfonction mitochondriale et un déséquilibre en calcium empêchant l’exocytose d’insuline.
Au final, les mutations dans des gènes de maintenance des télomères prédisposent à une perte de cellules souches et à des dérégulations cellulaires, qui sont à l’origine de diverses dysfonctions tissulaires. Malgré cela, de telles prédispositions génétiques ne suffisent pas à expliquer l’hétérogénéité des téloméropathies, notamment sur les différents âges d’apparition des premiers symptômes, et sur les différents niveaux de sévérité.
La longueur des télomères est héréditaire
Les familles portant une mutation dans un gène de maintenance des télomères ont la particularité de manifester une anticipation génétique. Autrement dit, au fil des générations, les descendants développent des symptômes plus précoces et plus sévères que leurs parents (12,37,41). Le taux d’anticipation génétique dans une famille dépend de l’impact du gène muté sur le raccourcissement accéléré des télomères (23).
La longueur raccourcie des télomères se transmet par les cellules germinales et n’est pas restaurée au cours du développement embryonnaire des descendants (12). Cela se démontre chez les enfants d’individus avec une téloméropathie due à un défaut des gènes TERT ou TERC, qui possèdent aussi des télomères raccourcis même s’ils n’ont pas hérité de la mutation (11,12). À ce jour, les données manquent pour savoir si ces individus possédant « seulement » des télomères raccourcis présentent plus de risques de développer certains phénotypes de téloméropathies. Une étude récente a tout de même démontré que des patients ayant hérité de télomères anormalement courts, sans avoir hérité des mutations familiales, avaient un risque accru de développer une fibrose pulmonaire idiopathique (46).
À noter que les tableaux cliniques sont extrêmement variables parmi les membres de la même famille. De manière générale, dans les familles prédisposées aux téloméropathies, les générations plus anciennes développent des symptômes plus tardifs, dans des tissus à renouvellement lent (ex.: fibrose pulmonaire, cirrhose cryptogénétique). À l’inverse, les générations plus récentes ont des phénotypes plus précoces, qui se manifestent particulièrement dans des tissus à renouvellement fréquent (ex.: insuffisance médullaire, anomalies cutanéomuqueuses) (11,26,47).
Finalement, dans le cas des téloméropathies, différents organes entrent en défaillance à différentes périodes de la vie. Cela s’explique d’une part par la variabilité du taux de renouvellement des différents tissus, impliquant un raccourcissement des télomères à une vitesse hétérogène dans l’organisme. De plus, les changements d’expression géniques dus à l’instabilité télomérique sont supposés avoir une influence variable suivant les tissus. Enfin, les organes peuvent être plus ou moins exposés à différents stress environnementaux accélérant le raccourcissement télomérique (11,12).
L’effet environnemental
L’influence de l’environnement sur le pronostic des téloméropathies est à ce jour peu décrite dans la littérature. Le principal phénomène connu concerne les familles portant une mutation de la télomérase, où les individus fumeurs sont plus à même de développer de l’emphysème pulmonaire9, tandis que les non-fumeurs présentent une prédisposition pour la fibrose pulmonaire idiopathique (48,49). De manière générale, il y a une interaction gènes-environnement forte, puisque dans les cohortes de patients symptomatiques, 65 à 75 % des patients rapportent une exposition à un aérocontaminant (tabac ou autre). La fumée de cigarette cause des dommages cellulaires s’additionnant à l’instabilité télomérique des cellules alvéolaires. Une hypothèse est donc que cet effet cumulé entraîne une mort cellulaire et donc la destruction des alvéoles, à l’origine de l’emphysème (48,49). À l’inverse, chez les non-fumeurs, l’épuisement progressif de cellules souches fonctionnelles et l’accumulation de cellules sénescentes stimuleraient le remodelage tissulaire et le développement d’une fibrose (11).
Davantage d’études sont nécessaires pour mieux comprendre l’effet de stress environnementaux sur l’évolution de différentes téloméropathies.
Traitements actuels des téloméropathies
À ce jour, il n’existe aucun traitement curatif des téloméropathies, car il est compliqué de compenser la perte de fonction d’une des protéines de maintenance des télomères et de soigner simultanément tous les organes atteints (poumon, moelle osseuse, foie, peau, cerveau, œil, tube digestif…). Les traitements sont purement symptomatiques, même si de plus en plus d’efforts sont effectués afin de dépister précocement les apparentés et de limiter l’exposition à des substances toxiques pouvant accélérer l’apparition de la maladie (24,30,50).
Pour les patients avec une atteinte hématologique telle qu’une aplasie médullaire sévère, le seul traitement est une greffe de moelle osseuse. Malheureusement, le pronostic de la greffe de moelle osseuse chez les patients téloméropathiques est mauvais, à cause de complications hépatiques et pulmonaires ainsi que d'une dysfonction du greffon (24). En effet, la greffe ne permet pas de corriger la mutation dans les cellules extra-hématopoïétiques, et un patient nécessitant une greffe pour sa survie immédiate aura souvent des anomalies sous-jacentes dans d’autres organes (24,30). En cas d’anomalies hématologiques, une autre option est d’administrer des androgènes, qui stimulent l’hématopoïèse. Un effet bénéfique des androgènes a été démontré chez des patients avec des téloméropathies, en particulier dans une série de patients avec une atteinte principale hématologique modérée (51).
Dans le cas d’atteintes pulmonaires et hépatiques, l’arrêt de la prise de toxiques tels que le tabac et l’alcool permet de ralentir l’évolution de la maladie. De même que pour la moelle osseuse, lorsque la défaillance hépatique ou pulmonaire devient sévère, la transplantation est envisagée. Mais une telle opération chirurgicale n’est pas sans risques, surtout chez des patients dont les capacités de régénération tissulaire sont limitées, et présentant souvent une déficience immunitaire liée à l’aplasie médullaire (30,50).
Une prise en charge psychologique est proposée chez les patients et leurs apparentés du fait du dépistage présymptomatique, de la présentation sévère de cette maladie et de la transmission génétique complexe (24,50).
Conclusion
Les progrès de la recherche sur la sénescence et les télomères ont permis de mieux comprendre certains phénotypes de vieillissement précoce, tels que ceux présents dans les téloméropathies. Les phénotypes du spectre de défaut de maintenance des télomères sont extrêmement variables, et dépendent à la fois de l’allèle muté, de la longueur héritée des télomères, et de l’exposition environnementale de l’individu. Malgré cette variabilité, plusieurs études ont démontré que le raccourcissement excessif des télomères et/ou leur instabilité suffisent à causer diverses dysfonctions tissulaires, évoluant en défaillance d’organes et vieillissement précoce.
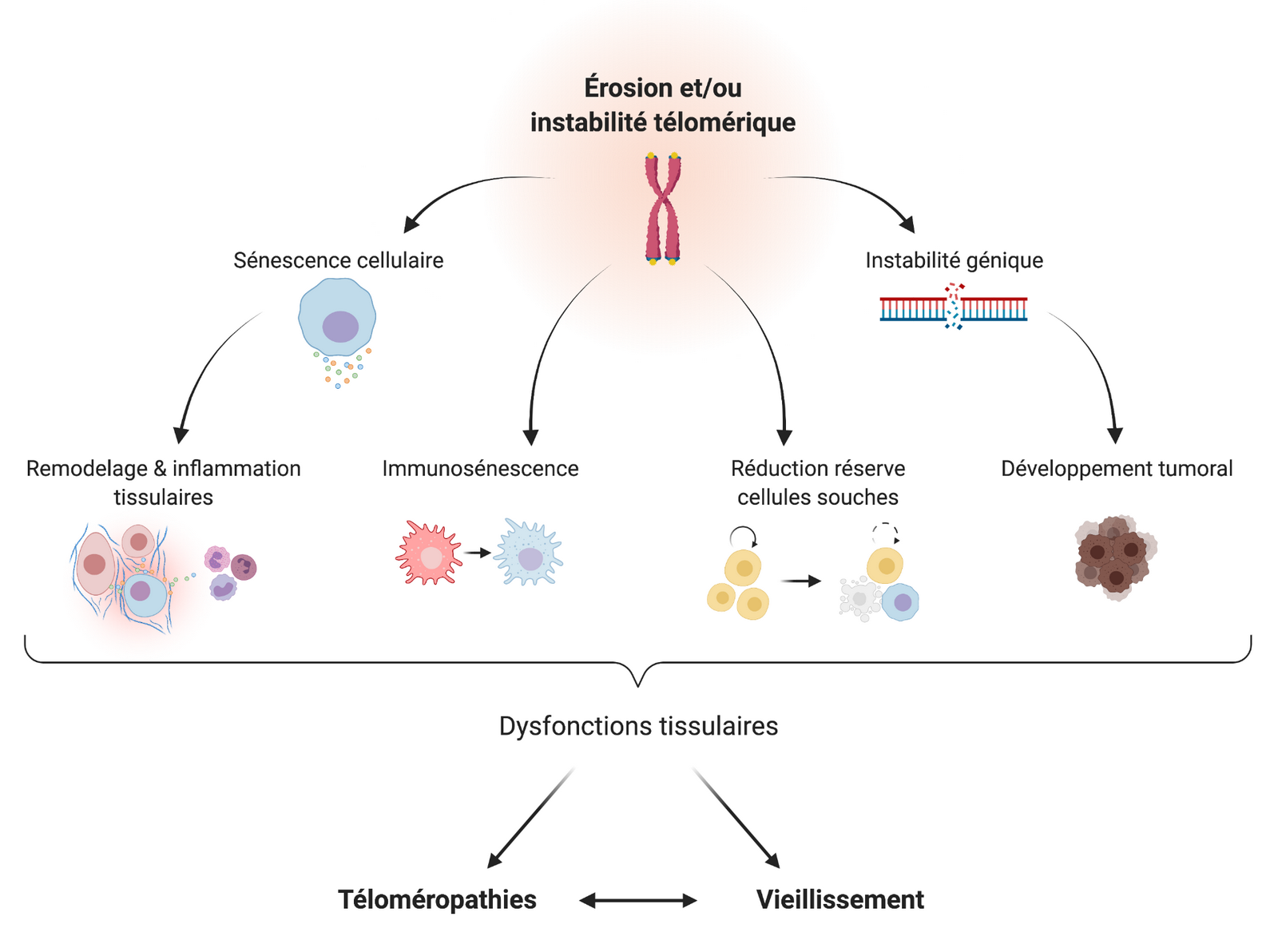
Dans le contexte d’une téloméropathie, la perte de fonction d’un gène de maintenance des télomères favorise l’érosion accélérée des télomères avec l’âge, voire une instabilité précoce de la structure télomérique (ex.: mutations du complexe shelterine). Cela engendre une augmentation de cellules sénescentes ou de mort cellulaire parmi les cellules prolifératives, notamment les cellules souches et les cellules du système immunitaire. Le déficit de régénération tissulaire et l’inflammaging accéléré précipitent la défaillance d’organes, à l’origine des divers symptômes de vieillissement prématuré que présentent les patients porteurs d’une téloméropathie. Schéma réalisé avec BioRender.com.
Références
1. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013 Jun 6 ; 153(6) :1194–217.
2. Dictionnaire médical de l’Académie de Médecine. In 2021. Available from : https://dictionnaire.academie-medecine.fr/
3. McHugh D, Gil J. Senescence and aging : Causes, consequences, and therapeutic avenues. J Cell Biol. 2018 Jan 2 ; 217(1) :65–77.
4. He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017 Jun 1 ; 169(6) :1000–11.
5. Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015 Jul ; 15(7) :397–408.
6. de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM, et al. Structure and variability of human chromosome ends. Mol Cell Biol. 1990 Feb ; 10(2) :518–27.
7. Blasco MA. Telomere length, stem cells and aging. Nat Chem Biol. 2007 Oct ; 3(10) :640–9.
8. Shay JW, Wright WE. Telomeres and telomerase : three decades of progress. Nat Rev Genet. 2019 May ; 20(5) :299–309.
9. Weinstein GD, van Scott EJ. Autoradiographic Analysis of Turnover Times of Normal and Psoriatic Epidermis**From the Dermatology Branch, National Cancer Institute, National Institutes of Health, Bethesda, Maryland. Public Health Service, U.S. Department of Health, Éducation and Welfare. J Invest Dermatol. 1965 Oct ; 45(4) :257–62.
10. Potten CS. Stem cells in gastrointestinal epithelium : numbers, characteristics and death. Crompton MJ, Dexter TM, Wright NA, editors. Philos Trans R Soc Lond B Biol Sci. 1998 Jun 29 ; 353(1370) :821–30.
11. Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012 Oct ; 13(10) :693–704.
12. Holohan B, Wright WE, Shay JW. Telomeropathies : An emerging spectrum disorder. J Cell Biol. 2014 May 12 ; 205(3) :289–99.
13. Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000 Oct ; 1(1) :72–6.
14. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell. 2013 Nov ; 155(5) :1104–18.
15. Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev Cell. 2014 Dec ; 31(6) :722–33.
16. Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011 Nov ; 479(7374) :547–51.
17. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016 Oct 28 ; 354(6311) :472–7.
18. Wu P, Takai H, de Lange T. Telomeric 3′ Overhangs Derive from Resection by Exo1 and Apollo and Fill-In by POT1b-Associated CST. Cell. 2012 Jul ; 150(1) :39–52.
19. Cooper G. Cell Proliferation in Development and Differentiation. In : The Cell : A Molecular Approach [Internet]. 2nd edition. Sunderland (MA) : Sinauer Associates ; 2000. Available from : https://www.ncbi.nlm.nih.gov/books/NBK9906/
20. Heller CG, Clermont Y. Spermatogenesis in Man : An Estimate of Its Duration. Science. 1963 Apr 12 ; 140(3563) :184–6.
21. Okamoto K, Seimiya H. Revisiting Telomere Shortening in Cancer. Cells. 2019 Jan 31 ; 8(2) :107.
22. R Bell D, Van Zant G. Stem cells, aging, and cancer : inevitabilities and outcomes. Oncogene. 2004 Sep ; 23(43) :7290–6.
23. Stanley SE, Armanios M. The short and long telomere syndromes : paired paradigms for molecular medicine. Curr Opin Genet Dev. 2015 Aug ; 33:1–9.
24. Haute Autorité de Santé (HAS). Protocole national de diagnostic et de soins (PNDS) APLASIES MEDULLAIRES ACQUISES ET CONSTITUTIONNELLES [Internet]. Haute Autorité de Santé ; 2019. Available from : https://www.has-sante.fr/upload/docs/application/pdf/2019-08/aplasie_medullaire_pnds__20190813.pdf
25. Armanios MY, Chen JJ-L, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase Mutations in Families with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2007 Mar 29 ; 356(13) :1317–26.
26. Armanios M, Chen J-L, Chang Y-PC, Brodsky RA, Hawkins A, Griffin CA, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci. 2005 Nov 1 ; 102(44) :15960–4.
27. Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, et al. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci. 2015 Apr 21 ; 112(16) :5099–104.
28. Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the Gene for Telomerase Reverse Transcriptase, in Aplastic Anemia. N Engl J Med. 2005 Apr 7 ; 352(14) :1413–24.
29. Kapuria D, Ben‐Yakov G, Ortolano R, Cho MH, Kalchiem‐Dekel O, Takyar V, et al. The Spectrum of Hepatic Involvement in Patients With Telomere Disease. Hepatology. 2019 Apr 10 ; hep.30578.
30. Savage SA, Alter BP. Dyskeratosis Congenita. Hematol Oncol Clin North Am. 2009 Apr ; 23(2) :215–31.
31. Dokal I, Vulliamy T. Telomerase Deficiency and Human Disease. In : Telomeres. 2nd Ed. Cold Spring Harbor Laboratory Press ; 2006. p. 139–61.
32. Savage SA. Dyskeratosis Congenita. In : Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. GeneReviews® [Internet]. Seattle (WA) : University of Washington, Seattle ; 2019 [cited 2021 Apr 1]. Available from : http://www.ncbi.nlm.nih.gov/books/NBK22301/
33. Kim N, Piatyszek M, Prowse K, Harley C, West M, Ho P, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994 Dec 23 ; 266(5193) :2011–5.
34. Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997 Nov ; 3(11) :1271–4.
35. Benyelles M, O’Donohue M-F, Kermasson L, Lainey E, Borie R, Lagresle-Peyrou C, et al. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Høyeraal–Hreidarsson syndrome. Hum Mol Genet. 2020 Apr 15 ; 29(6) :907–22.
36. Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, et al. Human RTEL1 deficiency causes Hoyeraal–Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013 Aug 15 ; 22(16) :3239–49.
37. Mason PJ, Bessler M. The genetics of dyskeratosis congenita. Cancer Genet. 2011 Dec ; 204(12) :635–45.
38. Sasa G, Ribes-Zamora A, Nelson N, Bertuch A. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet. 2012 May ; 81(5) :470–8.
39. Vulliamy T, Beswick R, Kirwan M, Hossain U, Walne A, Dokal I. Telomere length measurement can distinguish pathogenic from non‐pathogenic variants in the shelterin component, TIN2. Clin Genet. 2012 Jan ; 81(1) :76–81.
40. Marrone A, Stevens D, Vulliamy T, Dokal I, Mason PJ. Heterozygous telomerase RNA mutations found in dyskeratosis congenita and aplastic anemia reduce telomerase activity via haploinsufficiency. Blood. 2004 Dec 15 ; 104(13) :3936–42.
41. Kannengiesser C, Borie R, Ménard C, Réocreux M, Nitschké P, Gazal S, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J. 2015 Aug ; 46(2) :474–85.
42. Gu P, Min J-N, Wang Y, Huang C, Peng T, Chai W, et al. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012 May 16 ; 31(10) :2309–21.
43. Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres : analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008 Nov 1 ; 112(9) :3594–600.
44. Beier F, Foronda M, Martinez P, Blasco MA. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood. 2012 Oct 11 ; 120(15) :2990–3000.
45. Guo N, Parry EM, Li L-S, Kembou F, Lauder N, Hussain MA, et al. Short telomeres compromise β-cell signaling and survival. PloS One. 2011 Mar 10 ; 6(3) : e17858.
46. Vis JJ, Smagt JJ, Batenburg AA, Goldschmeding R, Es HW, Grutters JC, et al. Pulmonary fibrosis in non‐mutation carriers of families with short telomere syndrome gene mutations. Respirology. 2021 Sep 27 ; resp.14145.
47. Parry EM, Alder JK, Qi X, Chen JJ-L, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011 May 26 ; 117(21) :5607–11.
48. Stanley SE, Chen JJL, Podlevsky JD, Alder JK, Hansel NN, Mathias RA, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest. 2015 Feb 2 ; 125(2) :563–70.
49. Alder JK, Guo N, Kembou F, Parry EM, Anderson CJ, Gorgy AI, et al. Telomere Length Is a Determinant of Emphysema Susceptibility. Am J Respir Crit Care Med. 2011 Oct 15 ; 184(8) :904–12.
50. Armando RG, Mengual Gomez DL, Maggio J, Sanmartin MC, Gomez DE. Telomeropathies : Etiology, diagnosis, treatment and follow‐up. Ethical and legal considerations. Clin Genet. 2019 Jul ; 96(1) :3–16.
51. Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, Chen C, et al. Danazol Treatment for Telomere Diseases. N Engl J Med. 2016 May 19 ; 374(20) :1922–31.