Les alkylants sont des anticancéreux très utilisés en chimiothérapie. Ils agissent par la formation de liaisons covalentes avec l’ADN, entravant ainsi les processus de réplication et de transcription, et entraînant la mort cellulaire. Cependant, différentes voies de réparation peuvent être activées afin de réparer les lésions et ainsi contrer l’action cytotoxique des alkylants. Il est donc important de comprendre ces mécanismes dans le but de développer des traitements plus efficaces.
De nos jours, les cancers constituent l’une des premières causes de mortalité dans le monde, avec plus de 10 millions de décès enregistrés en 2020 1. Caractérisés par une prolifération incontrôlée des cellules, les cancers sont déclenchés par des mutations aléatoires ou héréditaires pouvant affecter différents types cellulaires et tissus biologiques. De plus, les tumeurs sont souvent composées de cellules ayant des propriétés différentes et pouvant évoluer vers des phénotypes plus ou moins résistants 2. L’hétérogénéité et le dynamisme des cellules cancéreuses constituent un véritable frein au développement de traitements efficaces.
La chimiothérapie est l’une des principales stratégies utilisées contre les cancers et vise à éliminer les cellules cancéreuses en prolifération. Découverte par accident durant la Seconde Guerre mondiale avec les agents alkylants, divers composés chimiothérapeutiques ont progressivement été développés pour d’abord cibler les maladies diffuses, telles que les lymphomes et leucémies, puis les tumeurs solides. De nos jours, la chimiothérapie est utilisée en tant que traitement principal, en tant que traitement néoadjuvant3 pour réduire la taille de la tumeur avant opération, ou bien en tant que traitement adjuvant, afin d’éradiquer les métastases. La chimiothérapie peut aussi être combinée à d’autres types de traitements anticancéreux (immunothérapie, radiothérapie, autres chimiothérapies…) afin d’améliorer leur efficacité 4.
Il existe actuellement 5 classes majeures de chimiothérapies anticancéreuses ayant des modes d’action différents : les antimétabolites, les agents alkylants, les inhibiteurs de topoisomérases, les antibiotiques anticancéreux et les poisons du fuseau mitotique (Tableau 1). Cet article se focalise sur les composés alkylants, qui sont la plus ancienne classe d’agents anticancéreux et sont donc très utilisés. Ils éradiquent les cellules cancéreuses en formant des liaisons covalentes (adduits) avec l’ADN, ce qui engendre des dommages au matériel génétique et provoque la mort cellulaire. Contrairement aux antimétabolites et inhibiteurs de topoisomérases, les alkylants agissent indépendamment du cycle cellulaire et peuvent donc être utilisés contre les cancers à croissance lente 5.
Cependant, malgré leur efficacité, l’utilisation des agents alkylant rencontre des obstacles majeurs. En effet, une dose prolongée peut provoquer des toxicités hématologiques importantes, ce qui peut dans certains cas entraîner des leucémies secondaires 6. De plus, les mécanismes biologiques de réparation de l’ADN sont à l’origine d’une résistance importante aux agents alkylants car ils limitent les effets toxiques de ces traitements. Il est donc essentiel de bien les comprendre afin de développer de nouvelles stratégies pour surmonter ces problèmes 7.
| Type d’agent chimiothérapeutique | Mode d’action |
|---|---|
| Antimétabolites | Bloquent le métabolisme (ex : réplication) en imitant les substrats biologiques |
| Agents alkylants | Forment des adduits avec les bases de l’ADN → lésions |
| Inhibiteurs de topoisomérase | Bloquent la modification de la topologie de l’ADN → dommages indirects à l’ADN → lésions |
| Antibiotiques anticancéreux | Intercalants de l’ADN, production d'espèces réactives de l'oxygène, oxydation du squelette sucre-phosphate de l’ADN |
| Poisons du fuseau mitotique | Inhibiteurs des microtubules → arrêt de la réplication |
Les alkylants
Les alkylants représentent la classe d’agents thérapeutiques la plus utilisée en chimiothérapie. Ils ont été découverts lors de la Seconde Guerre mondiale lors d'un accident provoquant une libération massive de gaz moutarde, qui avait des toxicités hématologiques retardées sur les soldats (aplasies médullaires et diminution des leucocytes). En 1942, la moutarde azotée, un dérivé du gaz moutarde, est utilisée pour la première fois dans le traitement contre les lymphomes 8. Aujourd’hui, il existe de nombreux types d’agents alkylants, principalement utilisés contre les cancers à croissance lente tels que les gliomes et les cancers hématologiques 9.
Les alkylants agissent en transférant un groupe d’alkyle10, le plus souvent un méthyle (–CH3), sur les atomes d’azote ou d’oxygène des bases adénine, guanine et cytosine. En fonction du nombre de sites réactifs qu’ils possèdent, ces agents peuvent former différents types de lésions : les alkylants monofonctionnels induisent des monoadduits sur un brin d’ADN, alors que les alkylants bifonctionnels peuvent former des adduits intrabrins, interbrins ou entre ADN et protéines (Figure 1). Ces modifications sont très toxiques pour la cellule car elles interfèrent avec des processus biologiques essentiels tels que la réplication et la transcription. De plus, elles provoquent des cassures double brin et des mésappariements, pouvant induire des mutations ou provoquer la mort cellulaire 11.

Néanmoins, les cellules disposent de cinq grandes voies de réparation de l’ADN pour contrer les différents types de dommages causés par les médicaments (Tableau 2, 1) :
- La réparation par excision de base (BER) est l’une des plus importantes car elle permet de corriger les modifications N7meG (méthylation sur l’azote 7 des guanines) et N3meA (méthylation sur l’azote 3 des adénines), qui représentent respectivement 75 % et 20 % des modifications causées par les alkylants.
- La réparation par excision de nucléotide (NER), moins fréquente, est impliquée dans la correction de lésions plus importantes, telles que les lésions intrabrins causées par des composés volumineux.
- Il existe également des systèmes de réparation directe, avec les enzymes MGMT ou ALKBH, qui permettent l’élimination du groupe d’alkyle sans cliver l’ADN.
- Si la base alkylée est répliquée de manière incorrecte, le système de réparation des mésappariements (MMR) peut corriger l’erreur, ce qui évite les mutations.
- Enfin, la recombinaison homologue (HR) permet de réparer les lésions double brin qui peuvent survenir suite à l’arrêt de la fourche de réplication au niveau de l’adduit.
| Système de réparation | Type d’alkylation |
|---|---|
| Réparation directe |
O6meG (réparation directe des O-adduits par l’enzyme MGMT) |
| Réparation par excision de base (BER) | N7meG, N3meA, N3meG (petites lésions) |
| Réparation par excision de nucléotide (NER) | Adduits volumineux |
| Réparation des mésappariements (MMR) | O6meG non réparés par MGMT |
| Réparation par recombinaison homologue (HR) | Lésions double brin |
Ainsi, les systèmes de réparation biologiques de la cellule peuvent limiter la toxicité des alkylants et conférer une résistance à ces médicaments. Nous allons étudier ces mécanismes à travers l’exemple de deux alkylants fréquemment utilisés : le témozolomide et le cisplatine.
Les systèmes de réparation et la résistance aux agents alkylants
Le témozolomide
Le témozolomide est un agent alkylant principalement utilisé chez les patients atteints de glioblastome, la tumeur cérébrale la plus fréquente chez l’adulte. En effet, cet agent est l’un des rares à pouvoir traverser la barrière hémato-encéphalique et est donc considéré comme le traitement standard pour ce type de tumeur. Néanmoins, environ 50 % des tumeurs ne répondent pas au traitement. Cette résistance est essentiellement provoquée par les systèmes de réparation directe, des mésappariements et par excision de base, qui sont les principales voies de réparation impliquées dans les lésions induites par le témozolomide 2.
L’enzyme MGMT
Le témozolomide agit par le transfert de groupements méthyles sur les positions N7 ou O6 des guanines et N3 des adénines (Figure 2). Cependant, alors que N7meG et N3meA représentent plus de 90 % des modifications induites par le médicament, c’est surtout la modification O6meG qui provoque la cytotoxicité et donc la mort cellulaire. En effet, la O6meG peut être mésappariée avec une thymine lors de la réplication puis prise en charge par le système de réparation des mésappariements, avec des conséquences mutagéniques et cytotoxiques (voir ci-dessous) 3.
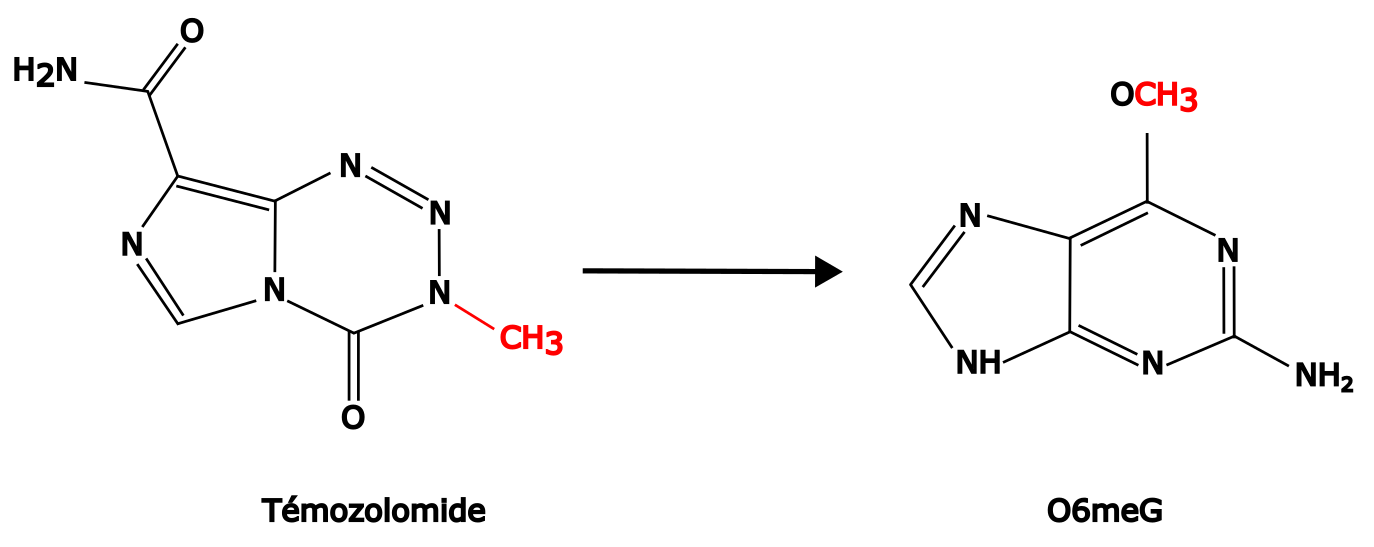
Les lésions O6meG sont principalement prises en charge par une réparation directe grâce à l’enzyme O6-méthylguanine méthyltransférase (MGMT). Cette enzyme peut directement cliver et récupérer le groupe méthyle, protégeant ainsi le génome de cette alkylation toxique (Figure 3).
Cependant, l’expression de l’enzyme, déterminée principalement par l’état de méthylation du promoteur du gène MGMT, peut varier selon les tumeurs et modifier la réponse au traitement. En effet, une hypométhylation du promoteur entraîne une surexpression de l’enzyme et donc une résistance accrue au traitement. Au contraire, une hyperméthylation provoque une diminution de la quantité d’enzyme exprimée, ce qui sensibilise les cellules au témozolomide. La méthylation de MGMT représente donc un facteur de prédiction de réponse au témozolomide, et peut être déterminé par pyroséquençage (technique de séquençage permettant d’obtenir le profil de méthylation du génome) lors de la biopsie. Les résultats obtenus permettent de différencier les traitements : les patients présentant une hyperméthylation de MGMT sont surtout traités par témozolomide, alors qu’en l’absence de méthylation, la radiothérapie est privilégiée. L’expression de MGMT peut cependant changer au cours du traitement par témozolomide et nécessite donc une surveillance pour adapter le traitement 1.
Le système de réparation des mésappariements (MMR)
Les cellules présentant une perte d’expression de MGMT peuvent utiliser une autre voie de réparation pour corriger les O6meG, la réparation des mésappariements. Cependant, alors que l’enzyme MGMT reconnaît directement l’alkylation, le système de réparation des mésappariements est activé suite à la réplication de l’ADN. En effet, les guanines O-méthylées (O6meG) sont reconnues en tant qu’adénine par l’ADN polymérase et appariées avec une thymine au lieu d’une cytosine. Ce mésappariement est d’abord identifié par le complexe MutSα (MSH2/MSH6), qui permet ensuite de recruter les enzymes impliquées dans le système de réparation des mésappariements afin de cliver le brin d’ADN et d’en retirer un fragment de plusieurs nucléotides, pour ensuite le résynthétiser et suturer le brin d’ADN. Cependant, ce système n’agit que sur le brin néosynthétisé et enlève donc la thymine, laissant la O6meG en place. Cela engendre un cycle futile persistant : à chaque nouvelle réplication, l’ADN polymérase apparie à nouveau une thymine à la guanine modifiée, ce qui réactive le système de réparation des mésappariements. Finalement, cette répétition provoque des cassures double brin, qui sont des lésions létales majeures pour la cellule (Figure 3). Par ailleurs, ces lésions double brin peuvent être réparées par recombinaison homologue, augmentant la résistance au médicament.
Le système de réparation des mésappariements est aussi susceptible d’être inactivé par mutagenèse lors du traitement. En effet, environ 25 % des glioblastomes acquièrent des mutations inactivatrices du gène MSH6, permettant ainsi la survie des cellules. Ces mutations sont notamment associées à la progression de la tumeur, car la présence de mésappariements induit une hypermutation génomique 2.
Ainsi, les tumeurs initialement MGMT−/−, et donc sensibles au témozolomide, peuvent voir leur système de réparation des mésappariements devenir défectueux pendant le traitement, ce qui favorise la survie des cellules mais également une instabilité génétique, caractéristique de nombreux cancers. La présence de mutations inactivatrices du système de réparation des mésappariements dans les tumeurs MGMT−/− peut donc être déterminée par séquençage génomique lors de la biopsie pour optimiser le choix du traitement.
La réparation par excision de base (BER)
Une dernière voie de réparation majeure, la réparation par excision de base, est impliquée dans la résistance au témozolomide. Ce système répare les adduits N3meA et N7meG, qui comptent pour environ 90 % des méthylations causées par le témozolomide 3. La réparation par excision de base permet de retirer les bases alkylées et de réparer les cassures simple brin qui surviennent suite à l’arrêt de la fourche de réplication sur les adduits. Les modifications N3meA sont reconnues par l’alkyladénine ADN glycosylase (AAG), qui clive le lien entre la base et le ribose, tandis que les modifications N7meG se dégradent spontanément, générant un site apurinique. Ces sites apuriniques sont ensuite reconnus par l’endonucléase AP (APE1) qui clive la liaison phosphodiester, générant une cassure simple brin qui est finalement réparée par une ADN polymérase (Figure 3) 4.
Les différentes protéines impliquées dans ce processus de réparation peuvent influencer la sensibilité au témozolomide. Notamment, une surexpression de l’enzyme APE1 favorise la réparation des sites apuriniques et donc la résistance au traitement. À l’inverse, une perte d’expression de APE1 accompagnée d’une surexpression de la glycosylase AAG sensibilise les cellules au témozolomide par l’accumulation d’intermédiaires toxiques tel que les sites apuriniques, qui ne sont pas réparés par APE1. Par ailleurs, cette sensibilité est accentuée par des mutations inactivatrices du gène codant l’ADN polymérase β, qui retire les 5’dRP (intermédiaires toxiques du système de réparation par excision de base formées lors de l’action d’APE1) et synthétise le nouveau brin d’ADN 56.
Ainsi, les enzymes AAG, APE1 et Pοlβ du système de réparation par excision de base peuvent être utilisées comme indicateurs de sensibilité au témozolomide lors de la biopsie, par séquençage génétique afin d’aiguiller le choix du traitement.
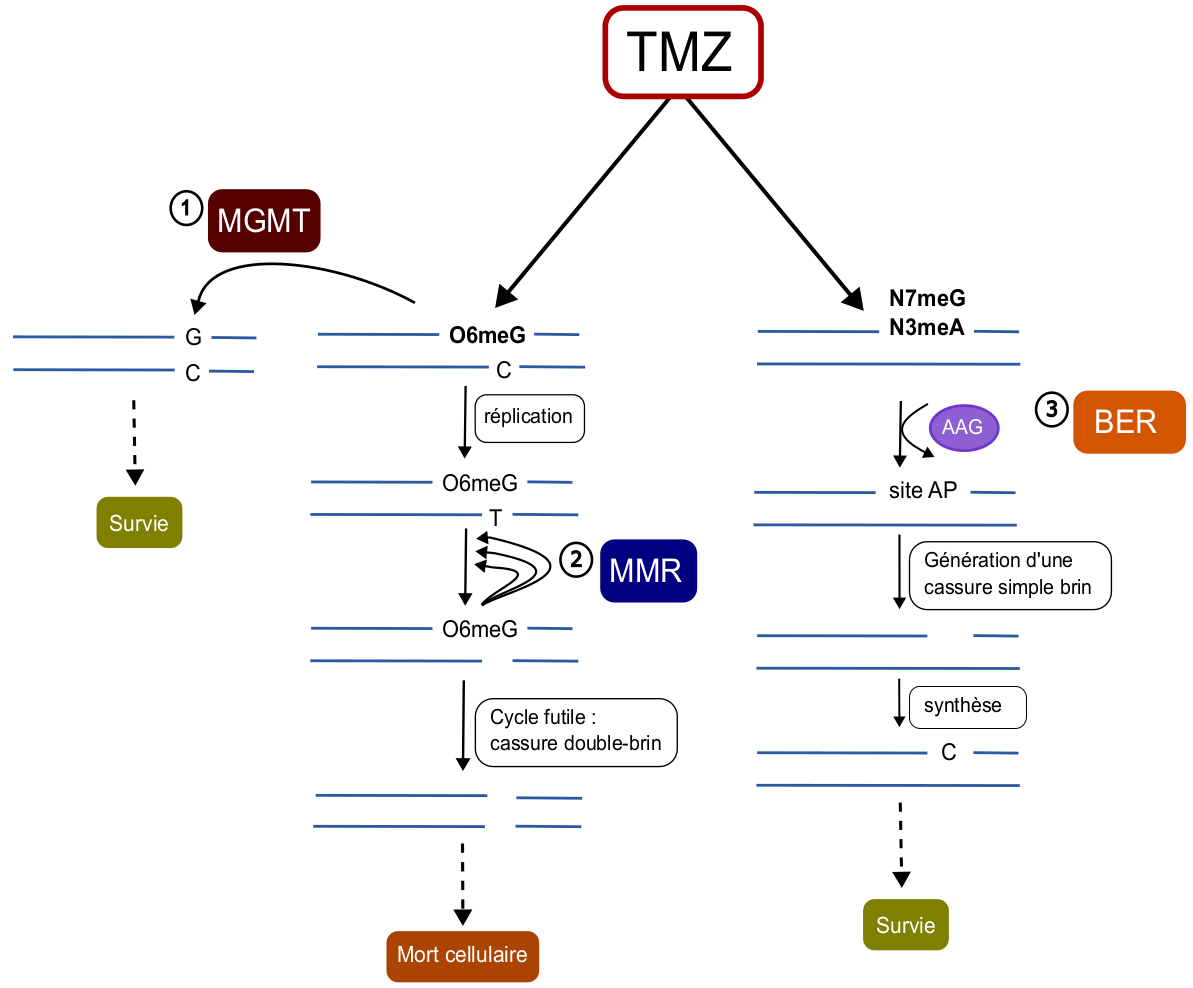
Les systèmes de réparation directe (MGMT) et de réparation par excision de base (BER) sont souvent surexprimés, car ils augmentent la survie de la cellule. À l’inverse, la réparation des mésappariements (MMR) est fréquemment perdue, car elle entraîne la mort cellulaire lors du traitement.
AAG : alkyladénine ADN glycosylase ; site AP : site apurinique ; TMZ : témozolomide.
Le cisplatine
Le cisplatine est un agent alkylant dérivé du platine qui, comme le témozolomide, est fréquemment utilisé en chimiothérapie. Initialement développé en tant qu’antibactérien en 1965, ses propriétés anticancéreuses ont ensuite été découvertes dans les années 1970 1.
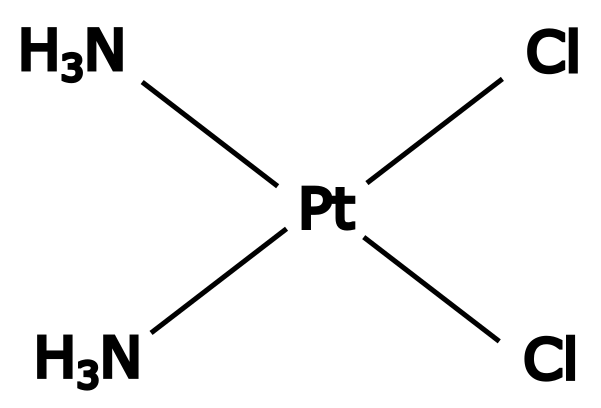
Le cisplatine exerce son action par la formation d’adduits interbrins ou intrabrins, en réagissant surtout avec l’azote 7 des bases puriques (adénine et guanine) (Figure 4). La formation d’adduits volumineux sur l’ADN entrave les processus de réplication et de transcription et engendre des cassures double brin toxiques pour la cellule. Cet agent est particulièrement réputé pour son efficacité dans les cancers des testicules, qui sont souvent caractérisés par une insuffisance de réparation de l’ADN. Cependant, les différentes voies de réparation de l’ADN peuvent contrer les effets du cisplatine et provoquer une résistance au traitement dans de nombreux cancers. Ainsi, les réparations par excision de nucléotide et par recombinaison homologue permettent de réparer les dommages liés au cisplatine, tandis que la synthèse translésionnelle permet la tolérance à cet agent. La réparation des mésappariements joue également un rôle dans la résistance au cisplatine lorsque l’adduit est répliqué de manière incorrecte, de la même manière que la résistance au témozolomide 1.
Réparation par excision de nucléotide (NER)
La réparation par excision de nucléotide est la principale voie de réparation activée en réponse au cisplatine. En effet, ce système est employé pour retirer les adduits volumineux sur l’ADN, tel que les adduits générés par le cisplatine, en supprimant directement le nucléotide modifié. Lors de ce processus, la modification est tout d’abord reconnue et les enzymes de réparation sont recrutées sur le site de la lésion. Le nucléotide endommagé est ensuite excisé, générant une cassure simple brin, qui est finalement réparée par synthèse et ligature des brins d’ADN (Figure 5).
Parmi les protéines du système de réparation par excision de nucléotide, ERCC1 est l’une des plus étudiées. En effet, il a été découvert qu’une surexpression du complexe ERCC1-XPF, impliqué dans l’excision du nucléotide, provoque une résistance accrue au cisplatine, notamment dans les cancers des ovaires. Au contraire, les patients ayant des cancers du poumons métastatiques avec une perte d’expression de ce complexe sont plus sensibles au médicament.
Le système de réparation par excision de nucléotide étant le principal mécanisme de résistance au cisplatine, ERCC1 représente donc un biomarqueur fiable qui peut être déterminé par immunohistochimie (méthode détectant la présence de protéines dans les cellules) lors des biopsies pour prédire la sensibilité des tumeurs au cisplatine et adapter le traitement des patients 1.
Réparation par recombinaison homologue (HR)
Dans les cellules dont le système de réparation par excision de nucléotides est défaillant, les dommages induits par le cisplatine génèrent des cassures double brin, les lésions les plus létales pour les cellules. Ces lésions, formées par l’arrêt de la fourche de réplication au niveau de l’adduit, activent la réparation par recombinaison homologue. Ce système repose sur une recherche d’homologie de l’ADN clivé, permettant de réparer les cassures double brin de manière précise et non mutagène.
Lors de ce processus, les lésions sont d’abord identifiées, entraînant la résection des extrémités des brins (nécessaire à la formation du segment d’ADN simple brin pour la recherche d’homologie). Des protéines Rad51 sont ensuite recrutées afin de recouvrir ces extrémités et guider les brins réséqués vers des segments homologues intacts, pour finalement permettre la synthèse et la ligature des nouveaux brins d’ADN 2 (Figure 5).
Ce processus de réparation est assisté par les protéines BRCA1 et BRCA2, qui favorisent le recrutement et l’activité des enzymes de réparation. L’importance de BRCA1/2 dans la sensibilité au cisplatine a été montrée par de nombreuses études, qui ont trouvé qu’une perte de ces protéines rend les tumeurs sensibles au médicament. Cependant, le traitement au cisplatine peut également provoquer des mutations restauratrices de BRCA1/2. Les patients atteints de cancer du sein BRCA-/- peuvent alors bénéficier d’un traitement au cisplatine plus efficace. Cependant, il est important de contrôler l’expression de ces protéines lors des biopsies pour éviter les rechutes 3.
Tolérance par synthèse translésionnelle
Enfin, les adduits intrabrins volumineux de cisplatine peuvent activer la synthèse translésionnelle. Ce mécanisme permet la tolérance de ces dommages, et évite les cassures double brin, l’instabilité génétique ou encore la mort cellulaire qui peuvent survenir suite à l’arrêt de la fourche de réplication sur les adduits. La synthèse translésionnelle fait appel à des ADN polymérases spéciales, les TLS Pol, qui permettent la réplication de fragments d’ADN portant des bases modifiées ou même des sites apuriniques grâce à leur site réactif plus large et à leur absence d’activité de relecture (Figure 5). Néanmoins, ces TLS Pol sont souvent infidèles et peuvent causer des mésappariements lors de la réplication, générant des mutations ponctuelles. Ces mutations sont caractéristiques d’une instabilité génétique et peuvent donc participer à la progression de la tumeur et être à l’origine d’une résistance accrue au cisplatine.
Ainsi, le développement d’inhibiteurs ciblant ce processus de tolérance permettrait un double effet anticancéreux, en sensibilisant la tumeur au cisplatine et en empêchant l’émergence de chimiorésistance 4.
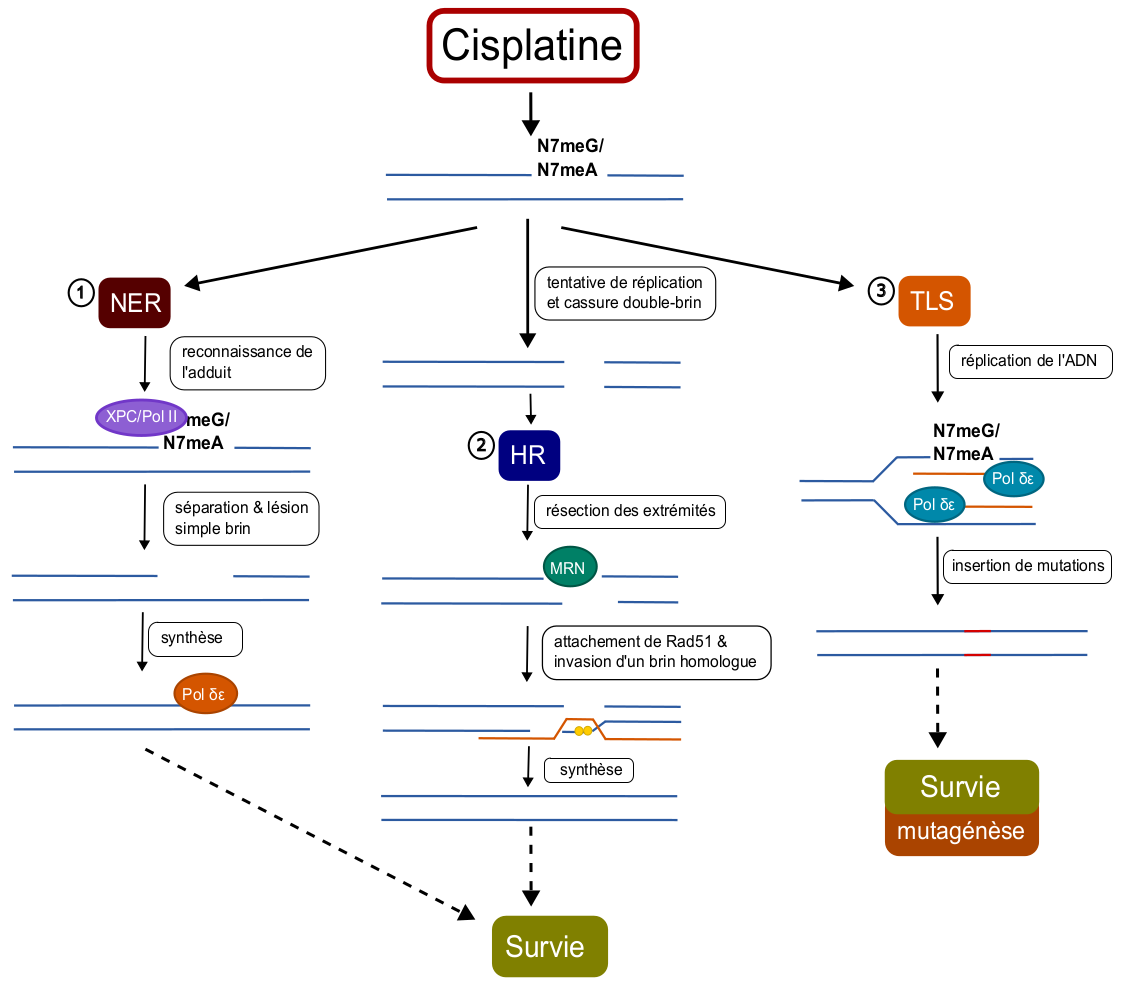
HR : réparation par recombinaison homologue ; NER : réparation par excision de nucléotide ; TLS : synthèse translésionnelle.
Conclusion
La chimiothérapie par alkylants est de nos jours un traitement standard utilisé contre de nombreux cancers. Ces agents agissent par la formation d’adduits sur l’ADN, qui entravent les processus biologiques et engendrent des lésions toxiques pour la cellule. Cependant, les différents systèmes de réparation de l’ADN de la cellule contribuent à la résistance aux alkylants, mais également à la progression de la tumeur. Il est donc important de déterminer quelles sont les voies de réparation actives dans les tumeurs lors des biopsies, afin d’adapter les traitements des patients et éviter les résistances. Enfin, la compréhension de ces mécanismes de résistance devrait également permettre de fournir, dans un futur proche, de nouveaux traitements anticancéreux plus sûrs et efficaces.