Caractéristiques de la sclérose latérale amyotrophique : symptômes, causes éventuelles aux niveaux cellulaires et moléculaires, difficultés du diagnostic et de la mise au point de traitements.

Jean-Martin Charcot est le premier médecin à avoir décrit la sclérose latérale amyotrophique en 1865.
La sclérose latérale amyotrophique (SLA), aussi connue sous le nom de maladie de Charcot, est une maladie neurodégénérative rare conduisant à une paralysie progressive puis à la mort des patients 3 à 5 ans après le diagnostic initial, lorsque les muscles de la respiration sont affectés. La SLA reste encore fatale et incurable. En France, sa prévalence est estimée à 1/25 000 personnes et 6 000 personnes vivent avec la maladie. Chaque jour, en moyenne, trois nouveaux cas sont diagnostiqués, ce qui en fait la plus fréquente des maladies rares du système moteur [1].
Les symptômes de cette maladie ont été décrits pour la première fois en 1865 et publiés en 1869 par le neurologue français Jean-Martin Charcot [3]. Le nom maladie de Charcot est de moins en moins utilisé, car il confond d’autres maladies décrites par le médecin [2]. De plus l’appellation SLA permet de décrire précisément les affections microscopiques et macroscopiques :
- sclérose : la dégénérescence des neurones moteurs laisse place à un tissu cicatriciel d’aspect dur et fibreux (sklêros signifiant dur en grec) ;
- latérale : correspond à l’affection des neurones et de leurs axones qui passent par le cordon latéral de la moelle épinière ;
- amyotrophique : le préfixe a renvoi à « l’absence de », myo signifie muscle et trophique est relatif à la nutrition des organes, tissus. Cela conduit donc à une fonte des muscles.
Ainsi, la SLA est due à la dégénérescence des neurones moteurs supérieurs (NMS, situés dans le cerveau et projetant vers le bulbe rachidien et la moelle épinière via des faisceaux de fibres latéraux) et les neurones moteurs inférieurs (NMI, situés dans le bulbe rachidien et la moelle épinière et projetant vers les muscles), conduisant donc à une paralysie et une fonte musculaire.
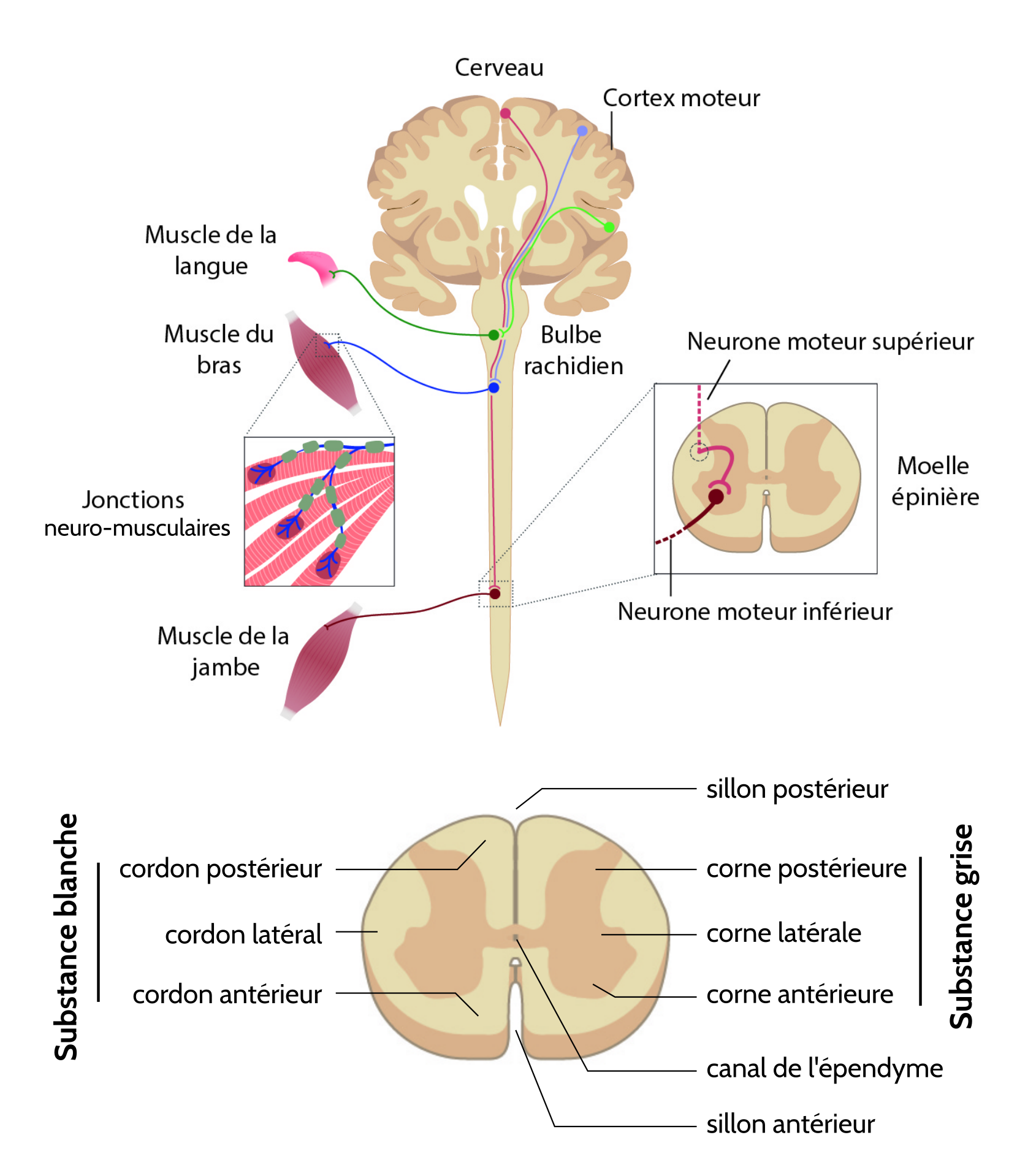
La SLA est due à la dégénérescence combinée des neurones moteurs supérieurs situés dans le cortex moteur et des neurones moteurs inférieurs situés dans la moelle épinière et le bulbe rachidien, entraînant une paralysie progressive des muscles.
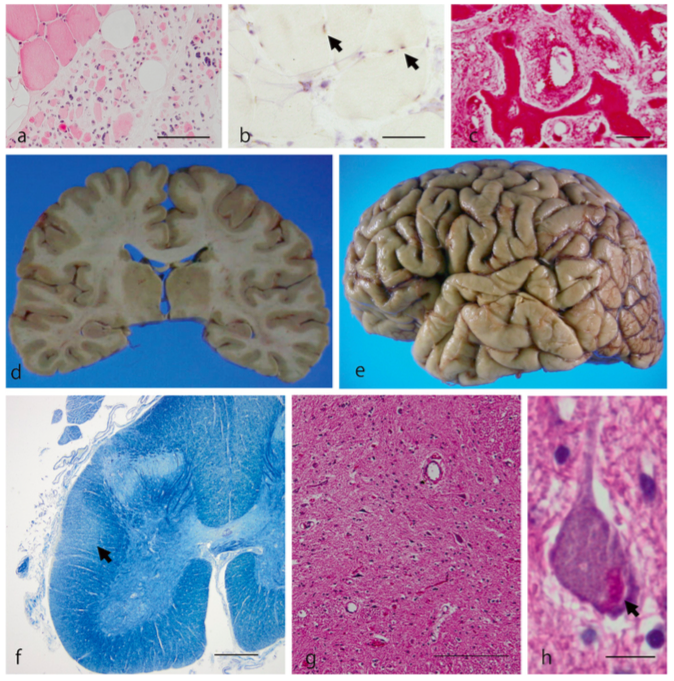
(a, b) Coupe de quadriceps atrophié présentant des agrégats de protéines (flèches). (c) Vertèbre entourant la moelle épinière avec des anomalies morphologiques. (d, e) Aucune atrophie cérébrale n’est à observer. (f) Dégénérescence des fibres du cordon latéral de la moelle épinière. (g, h) Perte des NMI de la moelle épinière, des inclusions protéiques sont présentes dans les neurones (flèches).
Malgré sa découverte, il y a plus de 150 ans, et d’intenses efforts de recherche, la SLA reste toujours incurable. Différentes raisons expliquent la difficulté à traiter cette maladie.
Ne pas confondre la sclérose en plaques (SEP) avec la sclérose latérale amyotrophique (SLA)
Les deux maladies sont dénommées sclérose en regard des affections du système nerveux, mais la SEP et la SLA sont deux pathologies bien distinctes. La SEP est une maladie auto-immune où le système immunitaire s’attaque aux cellules de l’organisme, et détruit la myéline qui est la couche protectrice se trouvant autour des axones des neurones. Les symptômes de la SEP peuvent être confondus avec ceux de la SLA et ont la capacité d’apparaître et de disparaître, ce qui n’est pas le cas pour la SLA où il y a une progression constante des symptômes. De plus, la SEP n’est pas une maladie fatale comme l’est la SLA.
La variabilité des symptômes
La maladie affecte un peu plus les hommes que les femmes (1,5 hommes pour 1 femme). Elle se déclare généralement entre 50 et 70 ans, avec une moyenne à 55 ans [2]. La dégénérescence des deux populations de neurones moteurs va engendrer plusieurs types de symptômes. Il existe une hétérogénéité considérable dans la manifestation des symptômes chez les patients atteints de SLA [4]. Ainsi, on classe les patients en fonction du lieu d’apparition des premiers symptômes [1].
-
Forme spinale : 1ers symptômes dans les muscles des membres (70 % des cas). Les symptômes résultants de l’atteinte des NMI contrôlant ces muscles sont des contractions ou secousses musculaires involontaires (fasciculations), accompagnés de crampes, ainsi qu’une faiblesse et une atrophie musculaires. L’atteinte des NMS conduit à une augmentation des réflexes, une hypertonie et une hyperactivité musculaire involontaire (spasticité). Ces différents symptômes amènent le patient à avoir des difficultés de coordination des mouvements, avec des gestes moins précis, un trouble de l’équilibre et de la marche avec ou sans chutes.
-
Forme bulbaire : 1ers symptômes dans les muscles de la face (25 % des cas). Le patient aura des difficultés pour parler, articuler ; sa parole devient lente, laborieuse et discontinue (dysarthrie spastique) ; sa voix change, devenant plus rauque, faible et nasillarde. Ce sont les signes de l’atteinte des NMS. Celle des NMI se manifeste par l’atrophie de la langue et du visage, accompagnés de fasciculations et d’une faiblesse musculaire. Le patient éprouve des difficultés pour déglutir (dysphagie), mâcher et bouger sa langue. Des fausses routes (passage de la nourriture ou de la salive dans les voies respiratoires) peuvent souvent arriver et conduire à des étouffements.
-
Autre forme rare : muscles de la respiration atteints en premier (5 % des cas). C’est la forme la plus dramatique car l’espérance de vie des patients est très faible ; ils doivent être mis très rapidement sous assistance respiratoire.
L’évolution de la maladie est progressive et va en s’aggravant avec une dispersion des symptômes vers les autres régions du corps, le patient va tendre vers une forme complète (bulbaire + spinale). La vitesse de progression de la maladie peut être très différente selon les patients et d’autres symptômes annexes sont susceptibles de se déclencher, comme une constipation, un amaigrissement général important, des problèmes liés à l’immobilité, des troubles du sommeil [1]. De plus dans 15 à 40 % des cas, les patients SLA vont développer des troubles cognitifs (désinhibition, problème de comportement social, apathie émotionnelle, trouble du langage) communs à ceux d’une autre maladie, la dégénérescence fronto-temporale (DFT)[6].
L’extrême variabilité des symptômes fait de la SLA une maladie neurodégénérative complexe et difficile à étudier tant le spectre des symptômes est large et varié. La compréhension de ces différentes formes phénotypiques est importante, car elle permet une meilleure prise en charge des patients, une meilleure formulation des pronostics, et de mieux sélectionner les patients pour les essais cliniques [5].

Paralysie, atrophie des muscles et troubles du langage sont les symptômes observables. Notons que cette forme de SLA est rare, car la progression des symptômes est très lente.
Un diagnostic par élimination
Il n’existe pas de test diagnostic définitif pour la SLA. Le diagnostic est basé sur l’évaluation des symptômes moteurs et leurs évolutions dans le temps. Le diagnostic de la SLA est fait par exclusion, car de nombreuses maladies du système moteur peuvent ressembler de près à la SLA [7]. Pour être atteint de SLA, le patient doit remplir les conditions suivantes :
- avoir des preuves cliniques de dégénérescence des NMI démontrées par électromyogramme et biopsie musculaire ;
- avoir des preuves cliniques de dégénérescence des NMS démontrées par électroencéphalogramme et imagerie médicale ;
- il doit y avoir une progression du syndrome d’une région à une autre du corps, déterminée par l’historique du patient ;
- absence de preuves pathologiques d’autres maladies pouvant expliquer les symptômes de la dégénérescence des NMI et des NMS.
Le diagnostic est difficile notamment à cause de l’hétérogénéité de la manifestation des symptômes. Chaque patient est différent et a son historique personnel. De plus, les patients consultent souvent à des stades tardifs de la maladie, car les premiers symptômes comme les crampes et la fatigue musculaire sont souvent minimisés et confondus avec d’autres affections. Aussi, si un patient se présente avec seulement des signes de l’atteinte des NMS, le diagnostic SLA pourra être confondu avec celui de la sclérose latérale primaire, une maladie où seuls les NMS sont affectés. Cependant, si ce patient développe plus tard des symptômes liés aux NMI, le diagnostic évoluera. C’est pour cela que bien souvent il faut plus d’un an pour poser le diagnostic de SLA [7].
Une absence de biomarqueur
Un biomarqueur est défini comme étant une caractéristique mesurable et quantifiable indicatrice d’un processus biologique normal ou pathologique. Un biomarqueur peut être une molécule (enzymes, protéines, hormones), des modifications de l’ADN, le score à un test de mémoire, la force musculaire, etc. Dans le domaine de la santé, un biomarqueur va pouvoir jouer plusieurs rôles. Il peut permettre le dépistage d’une maladie, le suivi de sa progression, refléter l’état du patient ou indiquer le bon fonctionnement d’une thérapie. Généralement, un biomarqueur est une molécule facilement dosable dans le sang ou les urines.
Dans le cadre de la SLA, on ne connaît pour le moment pas de bon biomarqueur [8]. La découverte d’un indicateur propre à la SLA permettrait d’améliorer la fiabilité et la rapidité du diagnostic. Dans l’idéal, il faudrait trouver des biomarqueurs spécifiques des différents stades de la maladie, qui permettraient aux cliniciens de prescrire les bons traitements aux bons moments. Cela permettrait également de constituer des groupes de malades plus homogènes pour les essais cliniques visant à développer des thérapies [10].
Pour l’instant, les efforts de recherche ont été vains, mais plusieurs biomarqueurs sont envisagés pour la SLA [9]. Des études sur le sang et le liquide céphalo-rachidien de patients SLA ont été réalisées, mais les résultats préliminaires restent à confirmer sur un plus grand nombre de patients. L’utilisation de biomarqueurs musculaires a également été envisagée, car le muscle est fortement endommagé par l’avancement de la pathologie. Cependant, la biopsie musculaire est une intervention invasive, ce qui limite les investigations longitudinales. De plus, le choix du muscle n’est pas forcément le plus pertinent, car selon les patients la maladie ne se déclare pas au même endroit et ne progresse pas de la même façon. Il est donc difficile de réaliser des études homogènes pour trouver un bon biomarqueur. Les nouvelles technologies en imagerie médicale permettent d’envisager de nouveaux indicateurs. En effet, grâce à l’IRM on peut maintenant voir plus en détail ce qu’il se passe dans le cerveau des patients et identifier des changements anatomiques ou même fonctionnels qui serviraient de biomarqueurs.
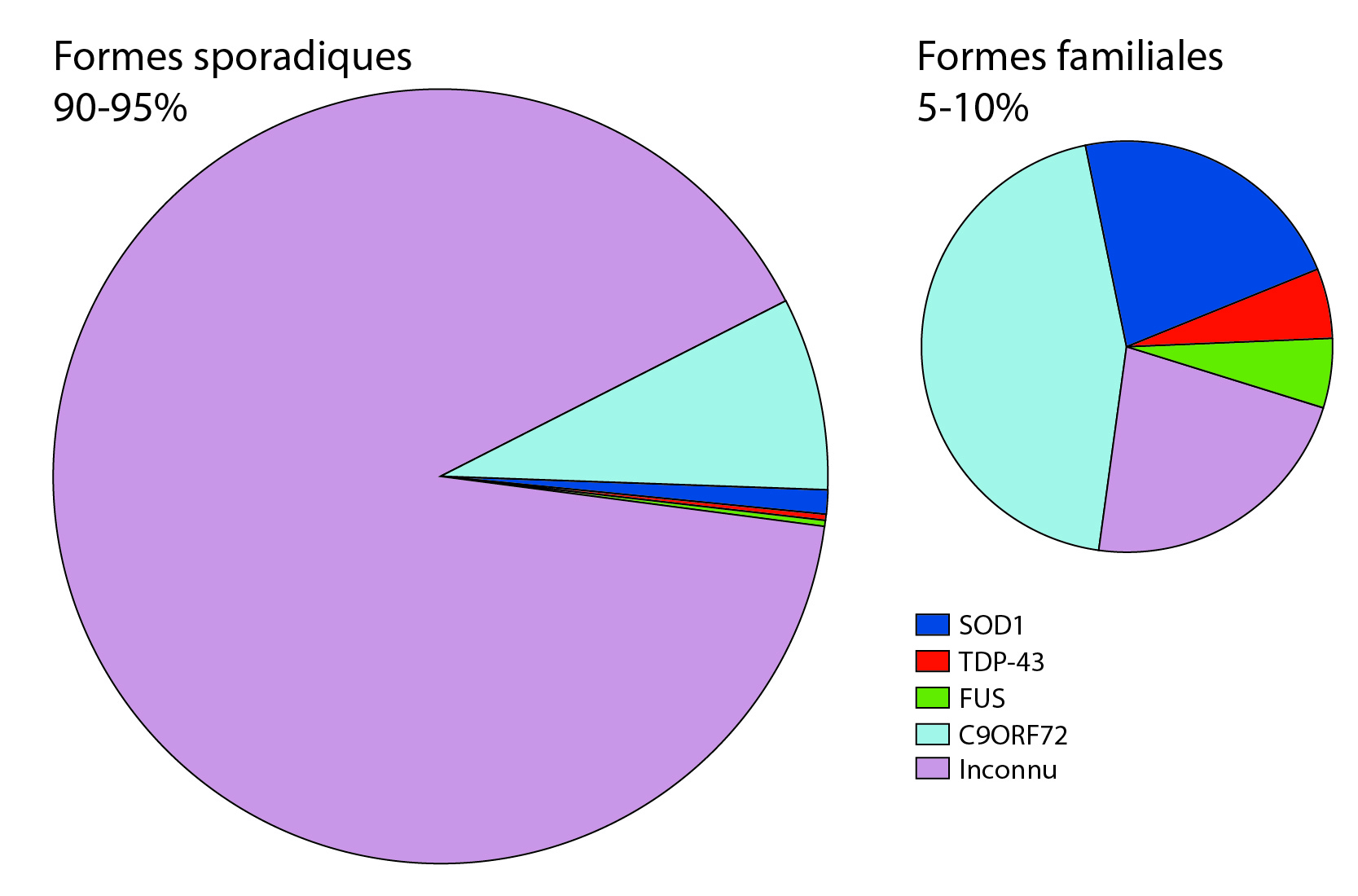
La SLA se distingue par deux formes, sporadique et familiale. Les causes de la SLA restent majoritairement inconnues. Seulement quelques mutations causant la pathologie ont été récemment découvertes.
Des causes variées encore mal identifiées
Les facteurs causant la SLA sont d’ordres génétiques et environnementaux, mais sont encore largement inconnus. Les patients sont séparés en deux groupes, ceux ayant une forme sporadique et ceux ayant une forme familiale [11].
Formes sporadiques
Elles représentent plus de 90 % des cas de SLA. Les causes de ces formes sont inconnues. De nombreux facteurs environnementaux ont été pointés du doigt sans qu’aucune étude ne puisse formellement confirmer leurs implications. Les facteurs environnementaux sont difficiles à mettre en évidence, c’est comme chercher une aiguille dans une botte de foin. L’exposome, tous les facteurs auxquels un patient est exposé au cours d’un temps défini, est immense. Aussi, les études sur les exposomes sont très coûteuses, car elles demandent de longues interviews de patients de façon longitudinale et une analyse fine des enregistrements. Ces études devraient être idéalement conduites sur au moins un millier de personnes pour faire ressortir des facteurs de manière significative. Et bien souvent, l’immensité de la tâche a conduit les chercheurs à commettre quelques fautes en prenant des raccourcis. De plus, certains paramètres environnementaux interagissent avec le fond génétique propre à chaque personne, et n’auront donc pas le même impact sur des personnes ayant des fonds génétiques très différents.
Plusieurs facteurs environnementaux ont été particulièrement étudiés dans la SLA, de façon plus ou moins arbitraire [11] : l’exercice physique, car les patients se présentant avec une SLA sont souvent de grands sportifs avec une silhouette athlétique et un indice de masse corporelle faible ; l’exposition à certains pesticides, métaux lourds ou produits chimiques a été mise en évidence sans que de réelles preuves soient démontrées ; le fait de fumer pourrait aussi être un facteur de risque de la SLA. Certaines corrélations ont aussi été avancées lorsque des études épidémiologiques ont montré que les footballeurs italiens ou les vétérans de la guerre du Golfe avaient plus de chance de développer une SLA.
Dans les formes sporadiques, des mutations génétiques peuvent apparaître spontanément chez les patients causant ainsi la SLA. Ces mutations sont aléatoires et non transmises à la descendance et peuvent se situer dans des gènes qui vont directement causer la maladie ou dans des gènes qui vont augmenter le risque de développer la SLA.
Formes familiales
Elles représentent environ 10 % des cas de SLA. Le patient présente alors des antécédents familiaux plaidant pour la transmission d’une mutation génétique causant la SLA [12]. Plus d’une dizaine de gènes ont été mis en évidence très récemment, mais d’autres restent encore inconnus. Il existe plusieurs types de transmissions géniques dépendants des gènes impliqués. Les types de mutations sont encore très discutés par les chercheurs. Parmi les gènes impliqués, on peut citer : SOD1, FUS, TDP-43, C9ORF72, etc.
Modèle gène-temps-environnement de la dégénérescence [11]
Il existe des interactions complexes entre les facteurs génétiques et environnementaux, qui s’influencent les uns les autres pour au final déclencher la maladie. Un autre facteur joue sur ces interactions : le temps. Plus les patients vieillissent et plus ces facteurs s’accumulent et augmentent le risque de développer la SLA. On parle alors d’un seuil de déclenchement de la SLA : lorsqu’il est franchi, la maladie se déclare et les symptômes commencent à apparaître. Ce seuil dépend donc des gènes hérités et des différentes expositions environnementales tout au long de la vie du patient. Selon les malades, les interactions entre les facteurs génétiques et environnementaux peuvent être différentes. Certains patients peuvent avoir un très mauvais fond génétique et une petite exposition suffira à déclencher la maladie. D’autres au contraire pourront avoir un bon fond génétique, mais vont être considérablement exposés, ce qui va engendrer la pathologie. Des situations intermédiaires peuvent aussi être envisagées dans ce modèle, avec un ou plusieurs gènes et un ou plusieurs facteurs environnementaux mis en jeu. Une chose est certaine, la SLA est une maladie neurodégénérative multifactorielle.
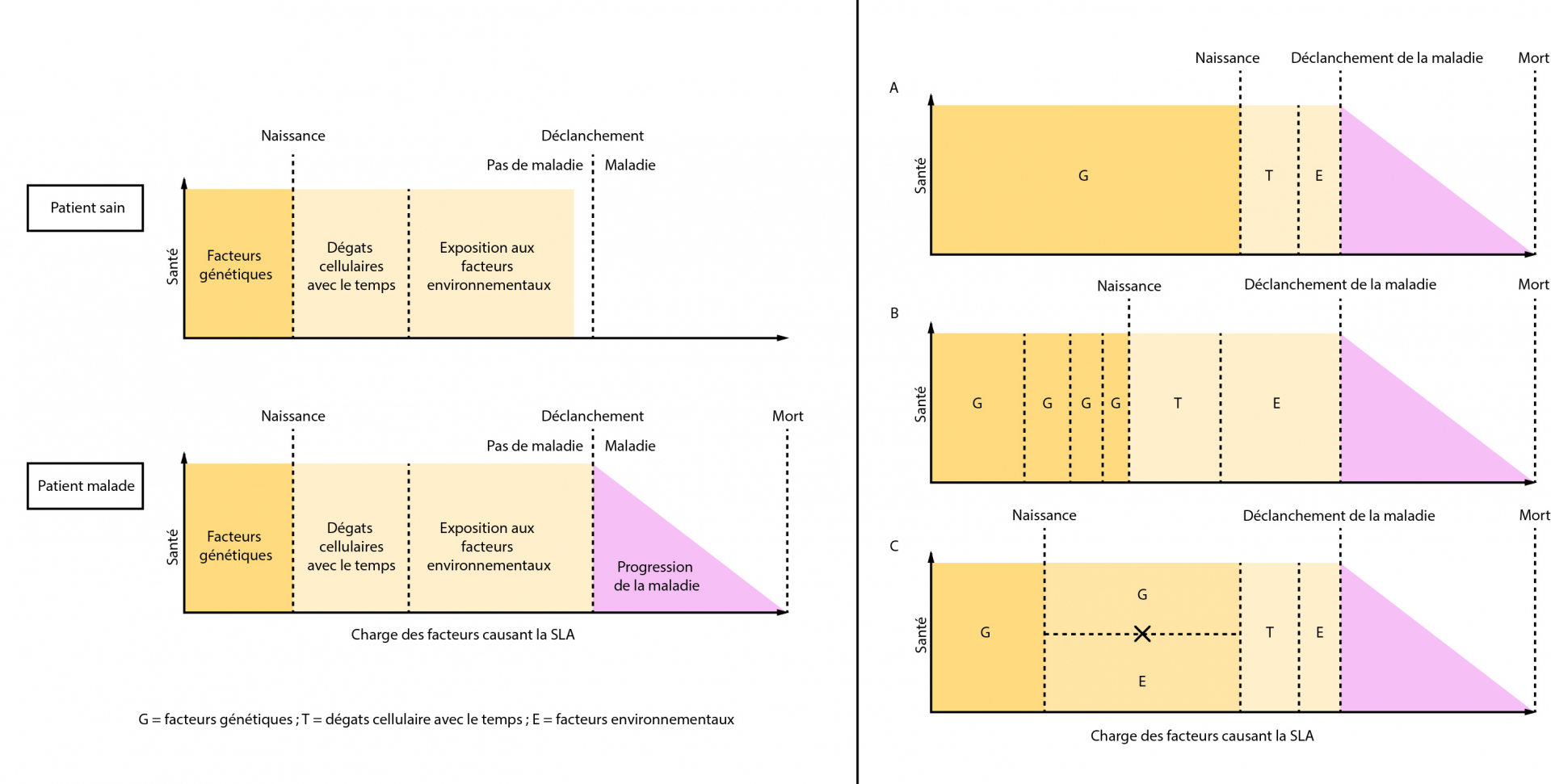
Le modèle gène-temps-environnement de la neurodégénérescence a été développé par Al-Chalabi et Hardiman pour expliquer le déclenchement de la SLA. La santé de l’individu est représentée sur l’axe des ordonnées et l’axe des abscisses correspond à l’accumulation des charges des différents facteurs entraînant la maladie. Lorsque le seuil de déclenchement est atteint, la maladie se déclare et l’état de santé diminue. Dans le modèle A, le fond génétique défavorable permet le déclenchement de la maladie sans nécessiter une grande exposition environnementale et un long temps. En B, plusieurs mutations se combinent avec une forte exposition environnementale sur une longue période de temps pour finalement déclencher la maladie. Le modèle C propose que la SLA est déclenchée grâce à une interaction spécifique entre des facteurs génétiques et environnementaux qui ont besoin d’être présents simultanément, dans une période de temps relativement courte.
Les mécanismes moléculaires commencent à être compris.
Les mécanismes moléculaires qui sous-tendent la dégénérescence des neurones moteurs supérieurs (NMS) et inférieurs (NMI) n’ont été découverts que très récemment et restent encore très débattus parmi la communauté scientifique. En effet, la SLA pourrait être causée par différents mécanismes aboutissant aux mêmes effets [6]. L’étude des mutations retrouvées dans les cas familiaux de SLA a permis de grandes avancées dans la compréhension des mécanismes moléculaires.
Comme dans de nombreuses maladies neurodégénératives, des agrégats protéiques sont retrouvés dans les neurones qui vont dégénérer. Ces agrégats vont induire un stress cellulaire en interférant avec les fonctions vitales de la cellule comme le transport intracellulaire, la dynamique du cytosquelette, le fonctionnement des mitochondries, etc. Chez les patients ayant une mutation dans le gène de la superoxyde dismutase 1 (SOD1), cette enzyme se retrouve agrégée dans le cytoplasme des NMS et des NMI. Ces agrégats font suite à une défaillance du système de dégradation des protéines, le système protéasome-ubiquitine. Certaines protéines participant à ce système, comme l’ubiquiline 2 (UBQLN2) peuvent être mutée et provoquer la SLA, indépendamment de la mutation du gène SOD1. Un deuxième système de dégradation des protéines peut également être affecté dans la SLA, on parle de l’autophagie. Cette voie permet de dégrader plus massivement des macromolécules et même des organites. Des protéines de ce système, telles que CHMP2B et VCP sont retrouvées mutées dans les cas de SLA.
On retrouve également beaucoup de mutations dans des gènes codant des protéines qui se lient à l’ARN, les plus fréquentes étant sur les gènes TDP-43 et FUS. Les protéines issues de l’expression de ces gènes sont normalement présentes dans le noyau des cellules. Mais dans les cas de SLA, elles se retrouvent agrégées dans le cytoplasme des neurones moteurs supérieurs et inférieurs. Cependant, l’origine de la toxicité de ces protéines est inconnue, elle pourrait être due à l’absence de la fonction originale de ces protéines, ou au gain d’une nouvelle fonction biologique qui serait alors toxique pour les neurones. Avec ces mutations, on observe un défaut de maturation des ARN messagers et des épissages alternatifs défectueux, conduisant à des protéines incorrectes.
L’implication des cellules gliales (astrocytes, microglies, oligodendrocytes) dans la pathogenèse de la SLA a été largement démontrée. Ces cellules forment l’environnement des neurones, permettent l’apport en nutriments et dioxygène et régulent l’homéostasie du milieu. Elles contribuent également à la dégradation des cellules mortes. Au cours de la progression de la maladie, les cellules gliales vont s’activer et participer au phénomène de neuroinflammation en sécrétant des molécules pro-inflammatoires qui peuvent être délétères pour les neurones. De plus, les cellules gliales peuvent contribuer à l’accumulation extracellulaire d’agrégats protéiques, d’une part en les sécrétant dans le milieu extérieur et d’autre part via un dysfonctionnement de leur action de nettoyage.
Le transport axonal se retrouve dérégulé dans les NMS et les NMI. En effet, les neurofilaments qui constituent le cytosquelette parcourant les axones sont désorganisés. Ainsi, les vésicules qui circulent le long de ces neurofilaments ne peuvent plus véhiculer les molécules le long de l’axone et cela induit une dégénérescence de l’axone.
D’autres hypothèses ont été étudiées comme par l’exemple l’excitotoxicité (neurotoxicité causée par une hyperexcitation) induite par le glutamate ou l’altération du métabolisme des lipides. La multitude des hypothèses proposées laisse penser qu’il existe un système cumulatif où différents mécanismes se combinent et s’accumulent jusqu’à la mort des NMS et des NMI.
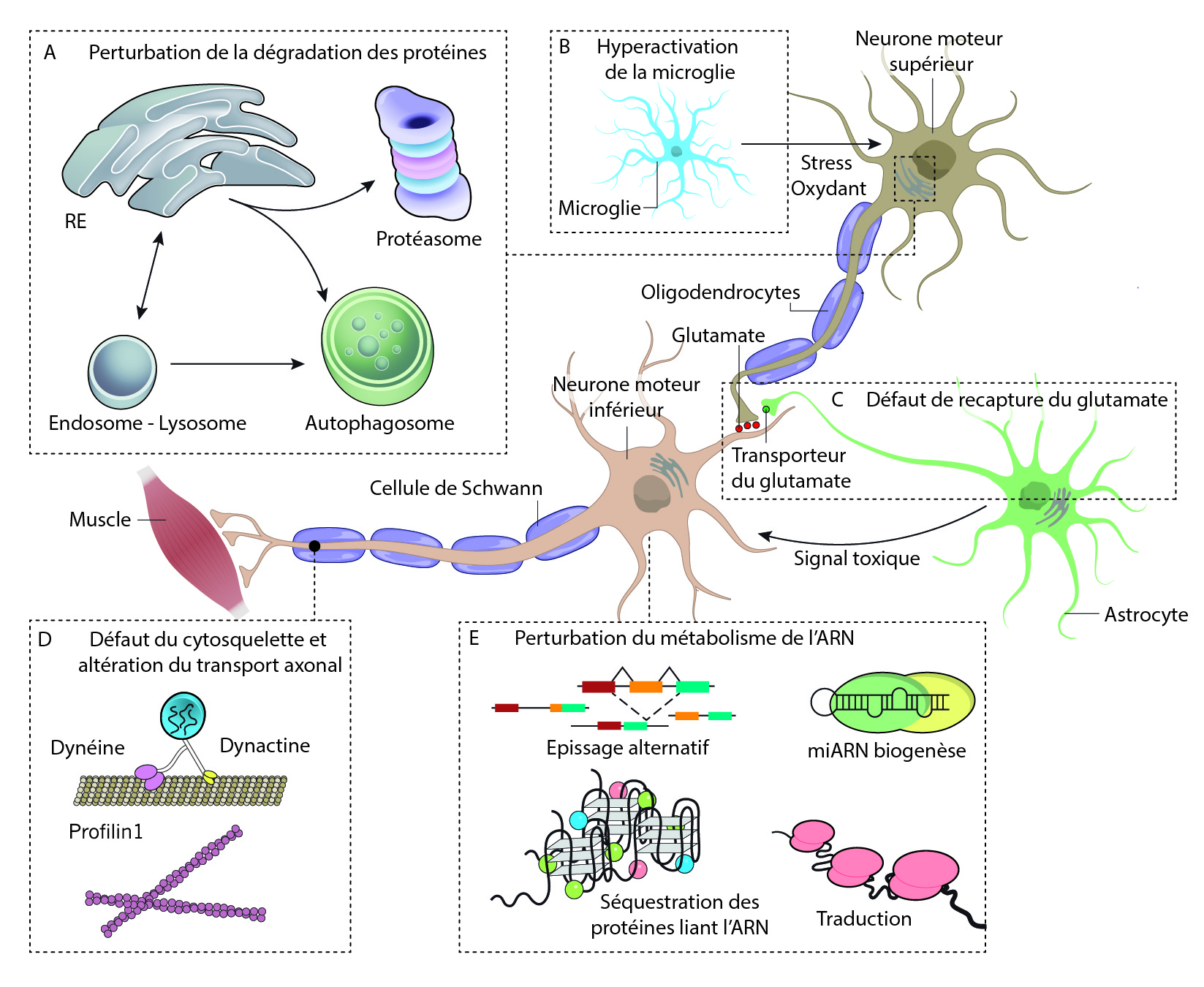
Plusieurs processus pathologiques ont été proposés, mais tous restent encore débattus.
Un seul traitement commercialisé
Les causes primaires de la SLA n’étant pas connues, il est très difficile pour l’industrie pharmaceutique et la recherche de développer un traitement efficace. Les modèles animaux ne rendant pas suffisamment bien compte la maladie, la transposition des traitements de l’animal vers l’homme reste une tâche ardue.
Pour soigner la SLA, un seul médicament peut être prescrit par un médecin ou neurologue : le Riluzole ou 2-amino-6-trifluoromethoxybenzothiazole [1]. C’est en 1995 que le médicament fut approuvé pour traiter la SLA ; avant cela, il était utilisé comme relaxant musculaire et anticonvulsivant (médicament utilisé pour lutter contre l’épilepsie). Ce médicament administré par voie orale de façon quotidienne permet d’augmenter l’espérance de vie des patients de deux à trois mois. Ces effets sont faibles, mais significatifs d’un point de vue statistique. Le Riluzole ne permet pas de sauver les NMS et les NMI de la mort qui les attend et l’on ne sait pas comment ce médicament agit sur le système nerveux ou sur d’autres cibles. Le Riluzole possède plusieurs cibles sur lesquelles il peut se fixer, il semblerait qu’il agisse sur le glutamate et inhibe son action excitotoxique qui peut causer la mort des NMS et des NMI. Cependant, cette hypothèse est encore très discutée et d’autres mécanismes seraient mis en jeu.
Le traitement de la SLA reste donc symptomatique [14] : les médicaments visant à atténuer ou plutôt soulager les symptômes comme les crampes, la fatigue, la spasticité, l’hypersalivation, etc. Lors de l’aggravation de la pathologie, les patients peuvent être mis sous aide respiratoire et dans le pire des cas une trachéotomie doit être pratiquée. Quand les muscles permettant la déglutition sont atteints, une sonde gastrique pour l’alimentation doit être implantée. D’autres symptômes peuvent être soignés par des spécialistes autres que le neurologue. Ainsi, le patient peut consulter un orthophoniste pour les problèmes de langage, un kinésithérapeute pour soulager les douleurs musculaires, enfin un soutien psychologique peut être réalisé par un psychologue.
La défaillance des modèles animaux
Pour étudier les maladies neurodégénératives comme la SLA, il faut combiner des études cliniques sur les patients et des études fondamentales sur des modèles animaux. Les études sur les patients permettent de décrire les symptômes et d’essayer de trouver les causes de la maladie. En revanche, il est difficile d’étudier les mécanismes sous-tendant la maladie chez l’homme. En effet, l’exploration est limitée par l’utilisation des techniques d’investigation, l’emploi de méthodes dites invasives ou ayant un risque pour le patient devant être limité au possible. Les méthodes non invasives seront privilégiées : prélèvement sanguin, techniques d’imagerie et, dans une moindre mesure, biopsie musculaire qui reste très douloureuse pour le patient. La collecte de ces informations est également soumise à la volonté du patient. Pour étudier plus en détail la SLA, les chercheurs ont dû créer des modèles animaux.
Un modèle animal est un animal non humain présentant une affection simplifiée que l’on retrouve chez l’homme et qui sert de modèle pour l’étude de cette affection. Tout modèle est une simplification utile et pertinente d’une réalité complexe. Dans l’idéal, le modèle animal reproduit les mêmes symptômes que l’affection, issue des mêmes causes et réagit de la même manière que l’homme aux traitements. Comme les causes de la SLA ne sont pas exactement connues, le développement de bons modèles animaux est difficile. Et la validation d’hypothèses doit bien souvent passer par l’étude conjointe de plusieurs modèles [9].
C’est la découverte des mutations dans les cas de SLA familiales qui a permis le développement des premiers animaux modélisant la SLA. Les modèles les plus utilisés sont les souris SOD1, créées en 1993. Ce sont des souris transgéniques qui surexpriment une forme mutée de la SOD1, mutation retrouvée dans 20 % des formes familiales [15]. Elles présentent des altérations motrices, puis leurs muscles se paralysent, les conduisant vers la mort en quelques semaines. On a là un modèle qui récapitule bien certains symptômes de la maladie mais qui a le défaut de présenter également des artefacts de transgenèse. D’autres modèles récapitulent d’autres symptômes par exemple les souris surexprimant des allèles mutants des gènes FUS ou TDP-43.
Différents animaux sont utilisés pour modéliser la SLA, par exemple le rat, le ver C. elegans, le poisson-zèbre, la mouche du vinaigre et même la levure S. cerevisiae, chacun ayant ses avantages et ses inconvénients. Sans oublier les modèles cellulaires de la SLA provenant d’animaux ou directement de patients, qui prennent une part de plus en plus importante dans la recherche.
Pour l’instant, les modèles de la SLA ont soulevé beaucoup d’interrogations concernant leur réelle pertinence. En effet, dans certains cas, les résultats obtenus chez les modèles animaux n’ont pas pu être confirmés chez les patients. Et certains résultats d’un même modèle diffèrent en fonction de l’institut de recherche où ils ont été obtenus. Globalement, il existe un manque de standardisation des méthodes scientifiques pour l’analyse de ces modèles [15].
Des essais cliniques peu concluants
Un essai clinique est une recherche biomédicale organisée et pratiquée sur l’homme en vue du développement de connaissances biologiques ou médicales [16]. Dans le cadre de la SLA, les essais cliniques ont pour but le développement de nouveaux médicaments et de nouvelles approches thérapeutiques. Plus d’une cinquantaine d’essais cliniques ont déjà été menés pour la SLA, sans aucun succès [17]. Pire encore, certaines molécules qui ralentissaient la progression de la maladie chez les modèles animaux ont accéléré la maladie chez les patients.
Plusieurs approches ont été tentées lors d’essais cliniques pour soigner les patients atteints de SLA. Ainsi, plus de dix mécanismes d’action ont été testés, ce qui reflète bien la complexité de la maladie et tous les efforts qui ont déjà été mis en œuvre. Les essais cliniques ont échoué principalement car les causes précises de la SLA sont encore inconnues, les modèles animaux ne sont pas parfaits et les méthodes scientifiques n’étaient pas toujours les bonnes. Dans certaines études, les molécules thérapeutiques ont été testées dès le stade présymptomatique sur les animaux. Or c’est impossible à faire chez l’homme, les patients arrivant bien souvent à des stades déjà très avancés de la maladie, ce qui peut expliquer les ratés des essais cliniques. Un autre problème important est l’hétérogénéité des patients qui rend les résultats compliqués à analyser. Par exemple, tous les patients ne prennent pas forcément la médication prescrite ou alors sont adeptes de l’automédication. Il existe donc beaucoup de variables pouvant venir influencer les essais cliniques et masquer un effet bénéfique du traitement. Aussi, le diagnostic difficile de la maladie fait que certains patients qui ne sont en réalité pas atteints de SLA sont inclus dans les études.
De nouveaux espoirs
En dépit de tous ces échecs, il existe de nouveaux espoirs et de nouvelles perspectives s’offrent au monde de la recherche pour la SLA. Depuis plus de vingt ans, les stratégies thérapeutiques se sont concentrées sur la recherche de molécules permettant la survie des neurones moteurs, sans grand succès. Le problème étant que quand les patients se présentent aux cliniciens, leurs neurones moteurs sont déjà bien atteints, et ces molécules ne stoppent pas les mécanismes pathologiques conduisant à la mort des neurones. Face à ce constat, les chercheurs tentent de nouvelles approches thérapeutiques comme la médecine personnalisée, où chaque patient est traité de façon individuelle. Cela est maintenant possible grâce aux cellules souches pluripotentes induites (CSPI, ou cellules IPS en anglais). Pour faire simple, le clinicien prélève des cellules de peau du patient, qui sont ensuite cultivées en laboratoire par des chercheurs, qui vont les faire revenir dans un stade indifférencié, le stade de cellule souche. Ensuite, en apportant des facteurs de différenciation, ils vont différencier les cellules souches en neurones moteurs et vont également corriger les mutations génétiques présentes dans ces cellules. Ces nouveaux neurones moteurs issus du patient pourraient ensuite être directement injectés soit dans le cerveau pour les neurones moteurs supérieurs, soit dans la moelle épinière pour les neurones moteurs inférieurs (NMI). Des chercheurs ont déjà créé des NMI à partir de cellules de peau de patients. Il reste maintenant à déterminer la sécurité de cette approche puis à la tester sur l’homme. D’autres thérapies cellulaires ont déjà été mises en place dans des essais cliniques comme l’injection de cellules souches mésenchymateuses dans la moelle épinière de patients. Ces cellules issues de la moelle osseuse vont sécréter autour des NMI divers facteurs qui vont les protéger de la mort cellulaire.
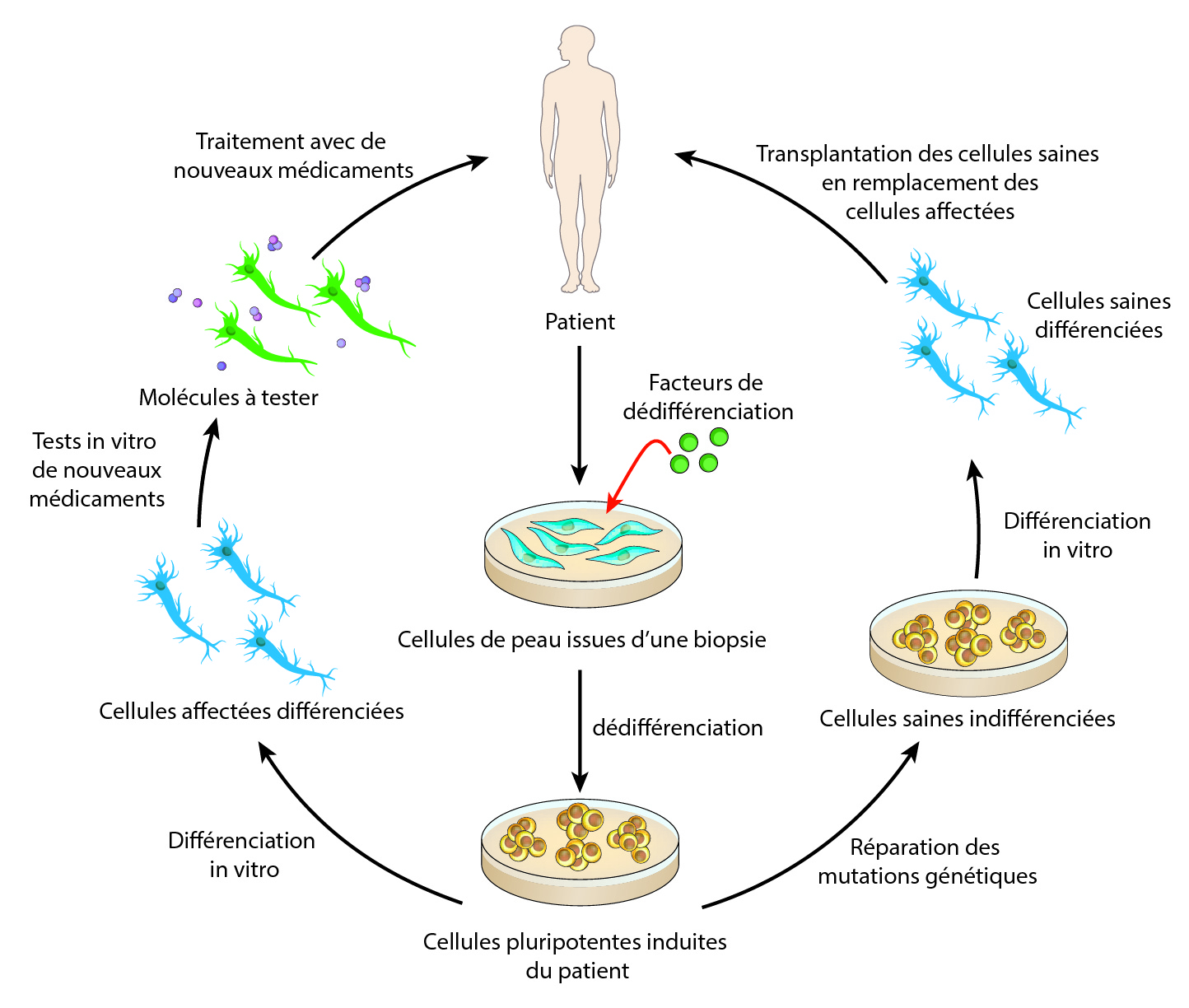
Des cellules de peau provenant du patient lui-même peuvent être différenciées en des neurones moteurs qui pourront, soit servir de modèle in vitro pour tester de nouveaux médicaments, soit être transplantés directement dans les zones affectées du système nerveux.
Une des problématiques de la recherche sur la SLA a été l’intense concentration des efforts sur les NMI et le muscle. Ces deux entités représentent plus de 90 % des résultats de recherche. Et les grands oubliés de la recherche sont les NMS qui représentent moins de 10 % des efforts de recherche aussi bien en recherche fondamentale que clinique. Cela est principalement dû à l’incroyable complexité de l’environnement dans lequel ils se trouvent, le cerveau, et de la toute la difficulté d’analyse que cela induit. Mais grâce au développement de nouvelles méthodes, de plus en plus de recherches sont faites sur les NMS et cela conduit à l’élaboration de nouvelles stratégies thérapeutiques, notamment l’utilisation de cellules souches neurales adultes. Nous savons maintenant que de nouveaux neurones sont générés de façon quotidienne tout au long de la vie de l’homme, au sein de la zone sous-ventriculaire, une zone située non loin des NMS. Ces cellules souches sont très malléables et il est possible de les différencier en NMS. Elles ont donc un intérêt thérapeutique immense pour tenter de soigner les patients atteints de SLA.
Le 5 mai 2017, l’agence américaine des produits alimentaires et médicamenteux a approuvé un nouveau médicament, le radicava [18] pour traiter la SLA, une première depuis 22 ans ! Ce médicament réduit le déclin des patients SLA de manière significative. La molécule active du radicava est un puissant antioxydant qui va limiter le stress oxydatif, connu comme étant responsable de la mort des neurones moteurs dans la SLA. Cette nouvelle constitue un nouvel espoir pour les personnes atteintes de SLA !
Références
- https://www.orpha.net/data/patho/Pub/fr/ScleroseLateraleAmyotrophique-FRfrPub106.pdf
- http://www.arsla.org/la-sla-cest-quoi/
- Charcot, J. M. & Joffory, A. Deux cas d’atrophie musculaire progressive avec lesions de la substance grise et des faisceaux antero-lateraux de la moelle epiniere. Arch. Physiol. Neurol. Pathol. 2, 744–754 (1869).
- Al-chalabi, A., Berg, L. H. Van Den, & Veldink, J. (2016). Gene discovery in amyotrophic. Nature Reviews. Neurology.
- Swinnen, B., & Robberecht, W. (2014). The phenotypic variability of amyotrophic lateral sclerosis. Nature Reviews. Neurology, 10(11), 661–670.
- Robberecht, W., & Philips, T. (2013). The changing scene of amyotrophic lateral sclerosis. Nature Reviews. Neuroscience, 14(4), 248–64.
- Turner, M. R., Hardiman, O., Benatar, M., Brooks, B. R., Chio, A., de Carvalho, M., Kiernan, M. C. (2013). Controversies and priorities in amyotrophic lateral sclerosis. The Lancet. Neurology, 12(3), 310–22.
- Bowser, R., Turner, M. R., & Shefner, J. (2011). Biomarkers in amyotrophic lateral sclerosis : opportunities and limitations. Nature Reviews. Neurology, 7(11), 631–638.
- Turner, M. R., Bowser, R., Bruijn, L., Dupuis, L., Ludolph, A., McGrath, M., Fischbeck, K. H. (2013). Mechanisms, models and biomarkers in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration, 14 Suppl 1, 19–32.
- Bakkar, N., Boehringer, A., & Bowser, R. (2014). Use of biomarkers in ALS drug development and clinical trials. Brain Research, 1–14.
- Al-Chalabi, A., & Hardiman, O. (2013). The epidemiology of ALS: a conspiracy of genes, environment and time. Nature Reviews. Neurology, 9(11), 617–28.
- Renton, A. E., Chiò, A., & Traynor, B. J. (2013). State of play in amyotrophic lateral sclerosis genetics. Nature Neuroscience, 17(1), 17–23.
- Bellingham, M. C. (2011). A Review of the Neural Mechanisms of Action and Clinical Efficiency of Riluzole in Treating Amyotrophic Lateral Sclerosis : What have we Learned in the Last Decade ? CNS Neuroscience & Therapeutics, 17, 4–31.
- Jenkins, T. M., Hollinger, H., & Mcdermott, C. J. (2014). The evidence for symptomatic treatments in amyotrophic lateral sclerosis, Curr Opin Neurol 27(5).
- Clerc, P., Lipnick, S., & Willett, C. (2016). A look into the future of ALS research. Drug Discovery Today, 21(6).
- http://ansm.sante.fr/Activites/Essais-cliniques/Qu-est-ce-qu-un-essai-clinique/(offset)/1
- Mitsumoto, H., Brooks, B. R., & Silani, V. (2014). Clinical trials in amyotrophic lateral sclerosis : why so many negative trials and how can trials be improved ? The Lancet Neurology, 13(11), 1127–1138.
- https://alsnewstoday.com/2017/05/08/fda-approves-radicava-first-new-als-therapy-in-20-years-and-cause-for-hope/
- Taylor, J. P., Brown, Jr, R. H. & Don Cleveland, D.W. (2016). Decoding ALS : from genes to mechanism. Nature, 539, 197-206.
- Ayaki, T. (2015). Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant Immunoreactivity of valosin-containing protein in sporadic amyotrophic lateral sclerosis and in a case of its novel mutant. Acta Neuropathologica Communications 2:172
- Robinton, D. A., & Daley, G. Q. (2012). The promise of induced pluripotent stem cells in research and therapy. Nature Reviews, 481, 995-305.