Une étude publiée au début de l’année 2022 dans la revue Nature [2] remet en question le dogme selon lequel les mutations se produisent de manière aléatoire dans le génome avant d’être sélectionnées [3]. En effet, ces travaux montrent que les régions fonctionnellement importantes du génome semblent comme protégées des mutations de novo, tandis que les séquences non transcrites ou non traduites mutent plus souvent. Dans ces régions très polymorphes, le poids des mutations dans l’évolution des séquences pourrait être plus important que celui de la sélection.
Les théories et modèles actuels d’évolution se basent sur l’hypothèse que les mutations arrivent de façon aléatoire, indépendamment de leurs conséquences, puis sont sélectionnées positivement ou négativement par l’environnement [4]. Une étude publiée en début d’année apporte des résultats pour le moins troublants, en montrant que le taux de mutation varie selon les séquences chez la plante modèle Arabidopsis thaliana. D’autres travaux avaient déjà étudié ce biais, que ce soit chez les procaryotes [5] ou les Eucaryotes [6], mais sans convaincre la communauté scientifique. En effet, ils ne proposaient pas de mécanismes pouvant expliquer leurs résultats, et utilisaient des méthodes controversées d’estimation du taux de mutation.
Pour étudier plus en détail ce biais de mutation, des chercheurs ont analysé plusieurs jeux de données génomiques de A. thaliana et ont quantifié sur chaque locus le nombre de mutations de novo, c’est-à-dire qui se produisent spontanément d’une génération à l’autre ou durant la vie de l’individu. Pour les identifier, ils ont étudié les accumulations de mutations sur plusieurs générations, en l’absence quasi complète de pressions de sélection, et comparé leur fréquence avec un modèle aléatoire.
Les résultats obtenus montrent que les mutations touchent moins les séquences codantes que les autres séquences du génome, et encore moins celles des gènes essentiels aux fonctions de base des cellules (Figure 1). De fait, au sein des gènes, les introns et séquences non traduites sont davantage susceptibles de subir des mutations que les exons. La fréquence des mutations dans les gènes est 58 % plus faible que dans les régions intergéniques, et 37 % plus faible dans les gènes essentiels que dans les autres gènes. Il avait bien sûr déjà été observé que les régions fonctionnellement moins importantes sont plus polymorphes, mais cette différence avait été interprétée comme le résultat de l’élimination des mutations délétères par la sélection naturelle après qu’elles se soient produites de façon aléatoire.
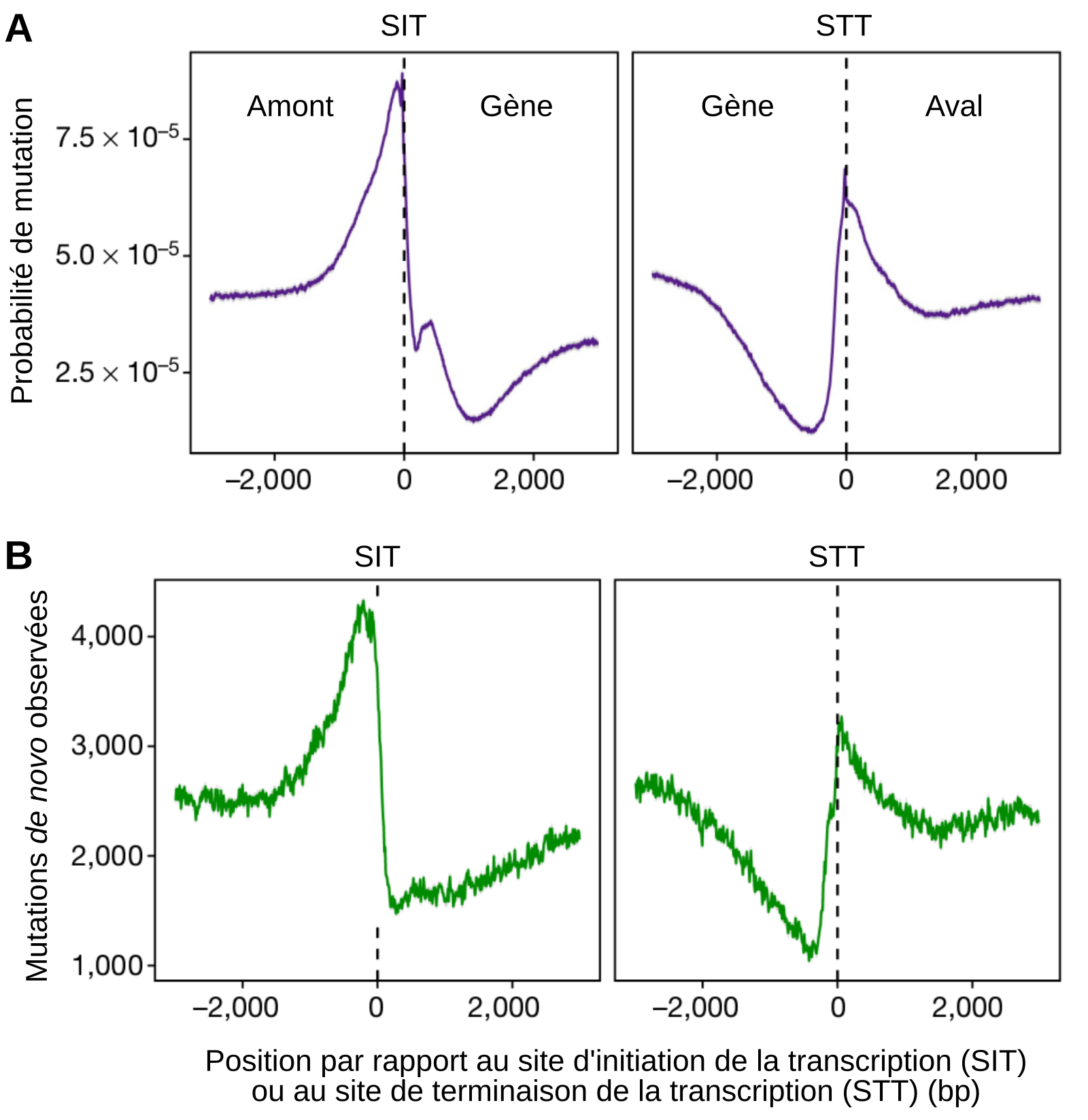
Probabilité de mutation et mutations observées en fonction de la position par rapport au site d’initiation (SIT) et au site de terminaison (STT) de la transcription. Deux types de mutations sont prises en compte : les SNV (single nucleotide variation), qui correspondent aux substitutions d’un nucléotide par un autre, et les indels, qui correspondent aux insertions et aux déletions.
A. Probabilité de mutation calculée à partir du modèle mis au point par les chercheurs, qui tient compte des caractéristiques épigénétiques locales.
B. Mutations de novo réellement observées, sur des lignées d’accumulation de mutations indépendantes de celles ayant servi à construire le modèle. Les résultats obtenus montrent une bonne concordance avec les prédictions du modèle.
Les chercheurs ont poursuivi leur étude en corrélant le taux de mutation observé à des caractéristiques épigénétiques (méthylation des cytosines, modifications post-traductionnelles des histones) et physiques (accessibilité) de la chromatine. Ils ont établi un indice prédictif de la probabilité de mutation en fonction de ces caractéristiques (Figure 2), dans un modèle qui parvient à reconstituer 90 % des résultats observés (Figure 1). Cela suggère que ces paramètres physiques et épigénétiques contribuent à moduler le taux de mutation le long du génome.
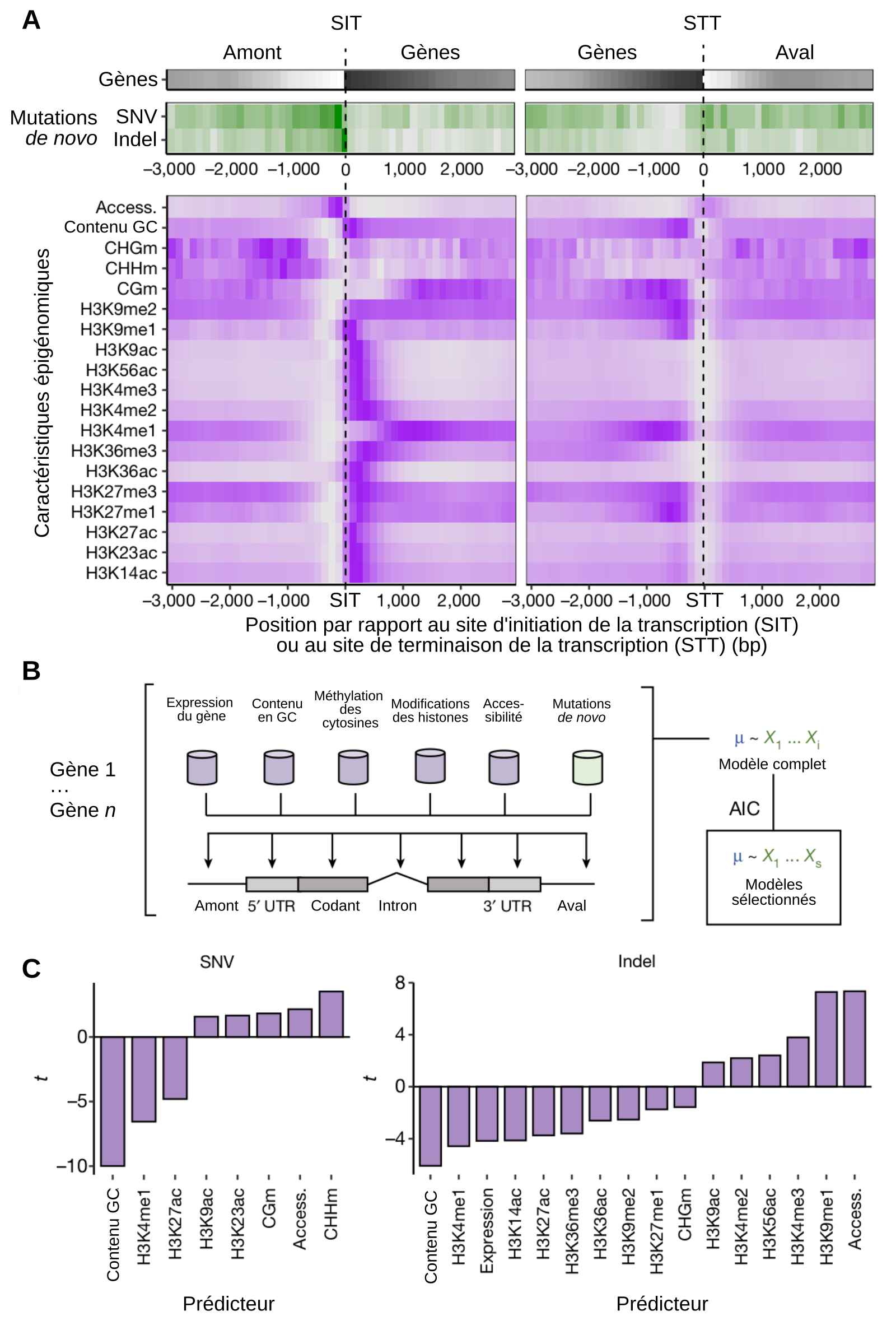
A. Densité des séquences transcrites (en noir1), des mutations observées (en vert), et de marques épigénétiques (en violet) dans tout le génome, positionnés relativement aux sites d’initiation (SIT) et de terminaison (STT) de la transcription. Deux types de mutations sont identifiés : les SNV (single nucleotide variation), qui correspondent aux substitutions d’un nucléotide par un autre, et les indels, pour insertion/déletion. Les notations CHGm, CHHm et CGm correspondent à des méthylations de cytosines dans différents contextes : C = cytosine, H = adénine, cytosine ou thymine, G = guanine. Les autres caractéristiques épigénomiques indiquées correspondent à des modifications d’histones. Il s’agit de mono, di ou triméthylations (me1, 2, 3) ou d’acétylations (ac) sur différentes lysines (K) de l’histone H3. Par exemple, H3K36me3 signifie que la lysine 36 de l’histone H3 est triplement méthylée.
B. Modèle prédictif des probabilités de mutations, établi à partir des données exposées en A. Le modèle est sélectionné parmi plusieurs modèles linéaires par comparaison des AIC (Akaike Information Criterion).
C. Valeurs statistiques liées au modèle prédictif. Une valeur positive de t indique une corrélation positive entre le type de mutation observé et une caractéristique épigénétique donnée. Par exemple, les méthylations des cytosines sur des séquences de type CHH (CHHm) augmentent la probabilité des mutations de type SNV, tandis que les méthylations de la lysine 4 de l’histone H3 (H3K4me1) diminuent la probabilité des mutations de type insertion-délétion.
1La densité des séquences transcrites est représentée avec un dégradé allant du noir au blanc. Si les régions en amont du site d'initiation de la transcription, et celles en aval du site de terminaison, ne sont pas entièrement blanches, c'est parce que ces régions contiennent également des séquences transcrites. Autrement dit, toutes les séquences transcrites d'A. thaliana ne sont pas forcément séparées par au moins 3000 pb.
À la lumière de ces travaux, il semblerait donc qu’un mécanisme de protection existe pour limiter le nombre de mutations sur les séquences fonctionnellement plus importantes. Il a été montré qu’un fort taux de GC tend à réduire le taux de mutations [7], alors que les marques épigénétiques, en fonction de leur nature, peuvent favoriser ou défavoriser les mutations [8]. De plus, on sait que les processus de réparation de l’ADN peuvent agir préférentiellement sur certaines régions [9]. Il est donc plausible que le biais de mutation observé soit également dû à une action des protéines de réparation.
La protection des régions fonctionnellement importantes aurait pour effet de raréfier les mutations sur ces séquences, mutations qui ont le plus généralement des conséquences délétères. Ainsi, le fait pour un organisme de disposer de mécanismes protégeant ses séquences importantes pourrait augmenter sa valeur sélective. Ces données remettent en cause le dogme selon lequel les mutations constituent une force évolutive aléatoire. Dans des régions non codantes, soumises à une faible pression de sélection mais dans lesquelles le taux de mutation est élevé, les mutations pourraient même représenter la force évolutive principale. Si ces résultats se retrouvaient chez d’autres espèces, cela remettrait en cause beaucoup d’analyses et de modèles qui avaient été réalisés jusqu’alors.
Références
[1] « More important genes mutate less », Detlef Weigel, Max Planck Institute for Developmental Biology, Nature Podcasts 19/01/2022
[2] Monroe et al, Mutation bias reflects natural selection in Arabidopsis thaliana, Nature 602, 101-105 (2022)
[3] Luria, S.E., Delbrück, M, Mutations of bacteria from virus sensitivity to virus resistance, Genetics, 28, 491-511 (1943).
[4] Futuyma, D. J. Evolutionary Biology 2nd edn (Sinauer, 1986).
[5] Martincorena, I., Seshasayee, A. S. N. & Luscombe, N. M. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485, 95–98 (2012).
[6] Hodgkinson, A. & Eyre-Walker, A. Variation in the mutation rate across mammalian
genomes. Nat. Rev. Genet. 12, 756–766 (2011).
[7] Wolfe, K. H., Sharp, P. M. & Li, W. H. Mutation rates differ among regions of the mammalian genome. Nature 337, 283–285 (1989).
[8] Weng, M.-L. et al. Fine-grained analysis of spontaneous mutation spectrum and frequency in Arabidopsis thaliana. Genetics 211, 703–714 (2019).
[9] Supek, F. & Lehner, B. Clustered mutation signatures reveal that error-prone DNA repair targets mutations to active genes. Cell 170, 534–547.e23 (2017)