L’ADN environnemental (ADNe) est une technique de pointe, de plus en plus utilisée au cours de ces dernières années. Elle consiste principalement en l’identification d’espèces à partir de l'ADN qu'elles laissent dans leur environnement. Cette méthode s’appuie sur des techniques classiques de biologie moléculaire (PCR, séquençage…). Bien qu’il existe de nombreuses limites, l’ADNe est utilisé dans des domaines variés allant de la génomique à l’écologie, en passant par la paléobiologie et l’évolution.
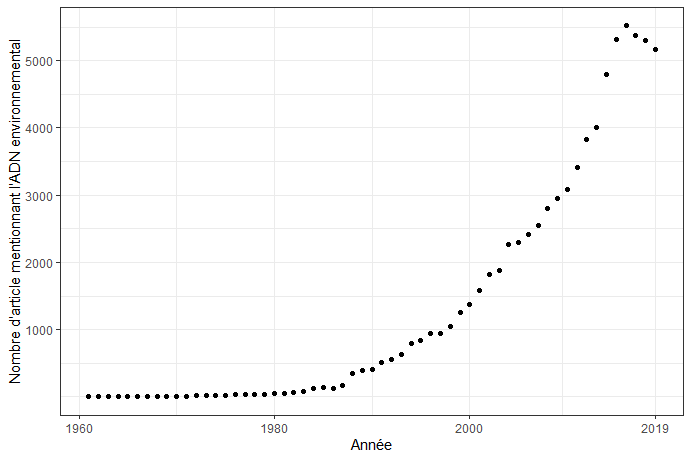
Graphique construit à partir des données disponibles sur Pubmed.
Ces dernières années, le nombre d’études utilisant l’ADN environnemental a augmenté très rapidement. Cette technique s’appuie sur l’analyse de l’ADN relâché par des êtres vivants, libre dans le milieu. L’expansion de cette méthode a été possible grâce à des progrès techniques dans le séquençage, qui réduisent ainsi son coût mais aussi le temps nécessaire à l’obtention et au traitement des données (Shokralla et coll. 2012). L’accès à une quantité très importante d’informations en appliquant des protocoles simples rend cette méthode utile dans différents domaines de la biologie et permet de répondre à plusieurs types d’objectifs.
De l’ADN dans l’environnement ?
Le principe de l’ADN environnemental repose sur le prélèvement et l’analyse de l’ADN présent dans un environnement. L’ADN étant le support universel de l’information génétique, l’analyse de ses séquences permet donc de différencier des individus et d’accéder à une multitude d’informations contenues dans celles-ci. Ainsi, en prélevant l’ADN que les individus laissent dans l’environnement, on peut indirectement détecter leur présence et généralement déterminer l’espèce à laquelle ils appartiennent. L’ensemble des taxons sont concernés par cette méthode, qui peut être appliquée dans différents types d’environnements (un océan, un échantillon de sol, un prélèvement de peau, le contenu d’un tube digestif ou les produits d’excrétion d’un animal).
Comment les espèces laissent-elles de l’ADN dans un environnement ?
Les individus interagissent en permanence entre eux et avec leur environnement. Ces interactions peuvent mener à la libération d’ADN dans le milieu. Cet ADN provient en général de cellules contenues dans des sécrétions, des excrétions (fèces ou urine), des tissus perdus lors du renouvellement cellulaire ou par blessure et également des cadavres d’individus morts. L’ADN libéré peut soit être toujours emprisonné dans les cellules, soit directement en contact avec le milieu après une rupture de la membrane plasmique des cellules. Lors des prélèvements, les scientifiques collectent les cellules et les molécules d’ADN libres.
État et utilisation de l’ADN
Une fois l’ADN dans l’environnement, sa préservation dans un état permettant des analyses dépend grandement et presque exclusivement des conditions environnementales dans lesquelles il se trouve. En effet, le temps de préservation peut varier de quelques minutes (dans un tube digestif par exemple) à plusieurs centaines de milliers d’années dans des sédiments ou dans la glace. Des études sur de l’ADN présent dans des sédiments ont permis de remonter jusqu’à 400 000 ans. Dans ce cas-là, l’ADN est évidemment très mal conservé mais est toujours source de nombreuses informations. C’est en effet de cette manière que l’existence d’ADN environnemental a été démontrée : la détection d’ADN de mammouth dans un échantillon de pergélisol a apporté une preuve indéniable de la conservation de l’ADN dans l’environnement (Willerslev et coll. 2003). L’ADN ne pouvait provenir d’une contamination par un individu actuel étant donné que cette espèce a disparu il y a 15 000 ans.
La stabilité de l’ADN extracellulaire est notamment compromise par les nucléases, enzymes dégradant l’ADN, qui sont, elles aussi, relâchées dans le milieu en cas de rupture des membranes. L’ADN est également très sensible aux fortes températures (supérieures à 60 °C) qui dénaturent la double hélice. Ainsi l’ADN sera d’autant mieux conservé que la température est basse et stable. Par ailleurs, pour des études concernant la reconstruction d’écosystème anciens, plusieurs sources d’ADN sont possibles : les sédiments terrestres ou aquatiques, la glace ou des sols congelés. Les sédiments se révèlent être très riches en ADN ; les sédiments marins sont de loin le réservoir le plus riche en ADN environnemental extracellulaire, ce qui s’explique par les conditions très pauvres en dioxygène, limitant la dégradation de l’ADN (Dell’Anno and Danovaro 2005).
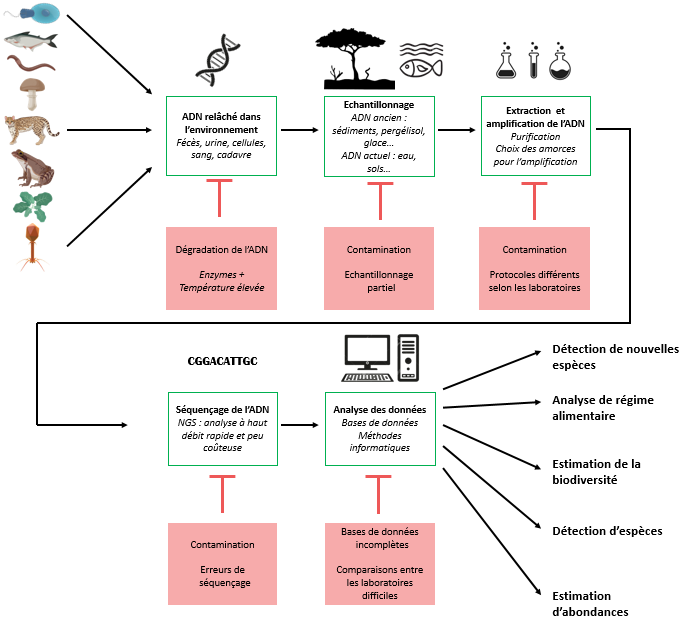
Du prélèvement à l’information apportée par l’ADN
Les manipulations pour analyser l’ADN environnemental sont globalement identiques mais peuvent varier légèrement selon la question que l’on pose. La majorité des étapes de ces manipulations nécessitent d’être très minutieux et peuvent être la source de nombreuses erreurs.
Échantillonnage de l’environnement
Différents types de milieux peuvent être échantillonnés : des milieux aquatiques (marins ou dulçaquicoles), des sols, des sédiments ou des milieux « biologiques » (comme un intestin par exemple). Les quantités prélevées varient selon le milieu et la précision que l’on veut obtenir : on peut aller de quelques millilitres d’eau à plus de cent litres. Les sédiments étant les milieux les plus riches en ADN environnemental, quelques grammes suffisent aux analyses. De même, pour les prélèvements terrestres (de sol par exemple) pour lesquels quelques dizaines de grammes sont prélevés (Bienert et coll. 2012). Dans tous les cas, les échantillons sont protégés par des agents conservateurs (éthanol ou solutions tampons spécifiques) (Bohmann et coll. 2014).
Extraction de l’ADN
Parmi tout ce qui a été prélevé, il faut désormais isoler et conserver uniquement les molécules d’ADN. Cette étape se fait en laboratoire grâce à des techniques de biologie moléculaire qui permettent de distinguer différentes molécules : suite à une lyse des cellules, les molécules d’ADN sont séparées des autres molécules (protéines par exemple) par solubilisation différentielle (méthode phénol/chloroforme), en utilisant des billes magnétiques ou des colonnes de centrifugation en silice sur lesquelles elles se fixent spécifiquement. Ces dernières techniques sont automatisables ce qui facilite le traitement à haut débit d’un grand nombre d’échantillons. Ainsi, tout l’ADN présent dans l’échantillon est isolé et purifié avant de poursuivre le protocole d’analyse.
Amplification de l’ADN
Une fois l’ADN extrait, on peut amplifier soit certaines soit toutes les molécules d’ADN en fonction de l’objectif de l’étude. Pour cela on utilise la technique d’amplification en chaîne par polymérase (Polymerase Chain Reaction, PCR), qui repose sur le processus de réplication d’ADN par une polymérase. Cette polymérase utilise des amorces, c’est-à-dire des courtes séquences d’ADN nécessaires à l’initiation de la synthèse de la séquence d’ADN par la polymérase. Ce sont les amorces choisies qui déterminent les séquences qui seront amplifiées et donc analysées : par exemple, des amorces spécifiques de gènes d’une espèce précise permettent l’amplification uniquement des molécules d’ADN de cette espèce. À l’inverse, l’utilisation d’amorces assez larges, qui ciblent des gènes partagés et conservés entre différentes espèces, au niveau du genre, de la famille ou même du règne, permettent d’étudier des taxons plus vastes. Classiquement, on utilise le gène COI (Cytochrome Oxidase I) pour les Animaux (Hebert et coll. 2003), les gènes rbcL et matK pour les Embryophytes (Plantes) (CBOL Plant Working Group 2009) et ITS pour les Champignons (Nilsson et coll. 2009).
Séquençage de l’ADN
Lorsque les séquences choisies ont été amplifiées, il est possible de les séquencer. Les méthodes employées ont énormément évolué depuis les années 1980. Les techniques de séquençage à haut débit (NGS, Next-Generation Sequencing) permettent d’obtenir rapidement et pour un coût faible la séquence des molécules d’ADN amplifiées.
Analyse des données
Une fois les séquences obtenues, on peut les comparer avec des séquences déjà connues qui sont répertoriées dans une base de données. Ce traitement des données se fait par des méthodes informatiques ce qui facilite et accélère l’analyse. C’est ainsi qu’il est possible de déterminer à quelles espèces appartenait l’ADN présent dans le milieu et donc quelles espèces étaient présentes dans le milieu. C’est l’une des nombreuses applications possibles de l’ADN environnemental.
Il semble important de remarquer que les progrès techniques ont permis le développement très important des recherches utilisant l’ADN environnemental. Par exemple, la deuxième génération des méthodes de séquençage a permis d’obtenir une grande quantité de séquences en un temps de plus en plus court et pour un prix de moins en moins élevé. De même, le développement d’outils informatiques puissants permet l’analyse rapide de toutes les molécules détectées dans l’environnement, ce qui était impossible à faire sans algorithmes adéquats. Enfin, la création de bases de données de séquençage de génomes a grandement contribué au développement de cette technique : plus le nombre de séquences connues sur les espèces est élevé, et plus il est facile d’attribuer une séquence d’ADN environnemental à une espèce à l’issu du protocole.
Problèmes rencontrés lorsqu’on travaille avec de l’ADN environnemental
Malgré l’existence de protocoles standardisés et reproductibles pour l’utilisation de l’ADN environnemental, les limites et problèmes sont nombreux et peuvent grandement influencer les résultats. Ces problèmes peuvent survenir lors de toutes les étapes. Nous en dressons ici une liste non exhaustive.
Échantillonnage et contamination
Le premier écueil qui se pose est celui de la représentativité de l’ADN qui est échantillonné par rapport aux communautés ciblées. Théoriquement, la probabilité de retrouver de l’ADN d’une espèce dépend directement de la densité d’ADN relâchée par cette espèce, qui dépend de son abondance. En particulier, une espèce peu présente risquera de ne pas être détectée. Cependant, ce biais d’échantillonnage existe dans toutes les méthodes d’estimation ou de détection de biodiversité. Ainsi, une espèce rare ne laissera que peu de molécules d’ADN qui auront peu de chance d’être échantillonnées : après analyse des données, l’espèce sera considérée comme absente du milieu. En lien avec les aléas de l’échantillonnage et de la préservation des molécules d’ADN dans un milieu hostile, il est possible de prélever beaucoup d’ADN d’une espèce rare et/ou peu d’ADN d’une espèce abondante, et donc de se tromper dans l’estimation des abondances des espèces.
Un autre problème concernant l’ADN à analyser est la contamination des échantillons. Il s’agit sans doute du problème le plus important, à l’origine de la détection de faux positifs. Les contaminations peuvent survenir à tout moment entre la collecte des échantillons et l’analyse des données. Le principal risque de contamination se fait dans les laboratoires d’analyse de l’ADN, qui traitent souvent des échantillons de localités différentes, et qui peuvent donc se contaminer entre eux. Ce phénomène est exacerbé par les nouvelles méthodes de NGS qui gèrent des millions de séquences simultanément. Afin d’éviter la saturation des analyses par de l’ADN indésirable, souvent humain, il est possible d’utiliser des bloqueurs de séquences spécifiques.
Erreurs d’amplification et de séquençage
En premier lieu, l’ADN libre dans l’environnement est vulnérable et donc souvent en mauvais état, ce qui peut compromettre la qualité des séquences amplifiées. Cependant il existe des méthodes permettant de réparer en partie les substitutions de bases causées par le milieu. De plus, les séquences étudiées sont courtes pour limiter les erreurs liées à la fragmentation de l’ADN.
L’échantillonnage d’ADN d’un milieu naturel s’accompagne de l’extraction de molécules du sol, telles que des inhibiteurs enzymatiques de polymérases, qui peuvent empêcher l’amplification de l’ADN (Matheson et coll. 2010). D’autres erreurs peuvent également se produire au cours de l’amplification des séquences, avec la formation de molécules chimériques (fusion de différentes séquences) ou l’apparition de mutations dans les séquences (Acinas et coll. 2005). Cependant, la majorité des erreurs de substitutions de bases surviennent lors du séquençage (Kunin et coll. 2010).
D’autres part, en fonction des protocoles utilisés (molécules, milieux, techniques de conservation, algorithmes employés pour traiter les données…), qui varient selon les laboratoires, les résultats de l’analyse d’un même échantillon peuvent être différents, ce qui pose le problème de la fiabilité de ces résultats et de la comparaison entre les laboratoires (Knudsen et coll. 2016).
Bases de référence
L’identification des espèces via leur ADN nécessite au préalable qu’une partie de leur génome soit déjà connue pour pouvoir faire un appariement exact. Il est donc crucial d’avoir à disposition des bases de données de références les plus complètes possibles. De plus en plus de banques de données génétiques se développent de manière coopérative, par exemple Genbank. Même si des efforts sont faits pour alimenter ces banques, elles restent encore très incomplètes, en particulier pour des taxons rares ou pour les espèces vivant dans des milieux peu connus. D’autre part, la majorité des séquences sont celles du COI (gène codant la sous-unité 1 de la cytochrome c oxydase) (Deagle et coll. 2006), le gène le plus utilisé pour la détermination d’espèces d’animaux, mais pas toujours fiable. En effet l’utilisation d’un seul gène pour différencier des espèces n’est pas optimale, la meilleure solution est d’analyser les génomes entiers, qui sont rarement retrouvés dans l’environnement. Les banques tendent à se compléter en présentant la totalité des génomes mitochondriaux ou nucléaires des espèces. Afin de confirmer l’identification d’une espèce, des combinaisons de gènes peuvent être utilisés, à défaut du génome entier.
Interprétation des résultats
La détection d’espèces à partir de l’ADN environnemental est, comme toutes les autres méthodes, incomplète. Autrement dit, dans une stratégie de ciblage où l’on cherche à détecter des espèces connues, seule une fraction de celles-ci pourra être détectée en pratique. Par ailleurs, même si la détection d’une séquence est correcte, son interprétation n’est pas simple : l’ADN reste le même quel que soit le stade de vie de l’individu (œuf, larve, adulte), qu’il soit vivant ou mort ou qu’il soit un individu hybride1. Ces différentes configurations peuvent avoir des conséquences complètement différentes en fonction des objectifs (la détection d’individus vivants pour de la conservation ; des individus jeunes indiquant le début d’une invasion d’une espèce non native…). D’autre part, la détection d’une séquence d’ADN à un moment et un lieu donnés ne signifie pas que l’individu était présent à ce moment et à cet endroit (par exemple, l’ADN relâché en milieu marin peut être transporté sur quelques kilomètres par les courants).
Quelles applications pour l’ADN environnemental ?
Analyse de régimes alimentaires
En analysant de façon ciblée l’ADN des contenus stomacaux d’animaux capturés ou de fèces, on peut directement avoir accès à un ensemble de données génétiques d’ADN provenant d’individus qui ont été ingérés et consommés. En détectant les espèces auxquelles appartiennent ces séquences, on peut en déduire le régime alimentaire de l’individu échantillonné. Cependant, l’ADN ayant été soumis à des conditions permettant la digestion et l’assimilation des molécules, il est souvent hautement dégradé (Pompanon et coll. 2012). Cet état de dégradation implique des ajustements dans les protocoles de traitements de l’ADN récupéré. À titre d’exemple, une étude en 2012 a permis d’identifier le régime alimentaire du chat-léopard, qui se compose de poissons, d’Amphibiens, d’Oiseaux et de Mammifères divers (Shehzad et coll. 2012).
Estimation de la biodiversité d’un écosystème
De nombreuses études montrent que l’utilisation de l’ADN environnemental permet une bonne estimation des différentes espèces (ou taxons) présents dans différents types d’environnements. En effet, cette méthode est efficace en milieux terrestre et aquatique. Des extractions d’ADN de sols de parcs africains permettent de détecter les grands Mammifères présents dans le parc (Andersen et coll. 2012). De même, une extraction de très faible quantité de sols de montagnes alpines (8 échantillons de 50 g), permet de détecter la présence de 80 % des espèces de vers de terre (les méthodes d’échantillonnage classiques ont le même pourcentage de détection) (Bienert et coll. 2012). Par ailleurs, la détection des espèces par la méthode de l’ADN environnemental est souvent plus efficace que les méthodes traditionnelles dans les milieux aquatiques (Watson et coll. 2005, Cantera et coll. 2019). En effet, la détection moléculaire par l’ADN environnemental inclut souvent des espèces cryptiques, cachées ou difficilement repérables à l’œil ou sur des vidéos (espèces de petite taille par exemple).
D’autre part, l’ADN prélevé dans les sédiments, le pergélisol ou les calottes glaciaires permet de déterminer les espèces ayant vécu dans ces milieux et de reconstruire, souvent partiellement, la biodiversité passée. De nombreux exemples utilisent cette méthode pour décrire la diversité passée de Plantes, Champignons, microorganismes ou de grands Animaux à partir d’ADN obtenus dans des milieux où celui-ci se conserve longtemps (sédiments, milieux froids) (Jørgensen et coll. 2012). Par exemple, des échantillons de carottes glaciaires du Groenland datés d’il y a 2 000 à 4 000 ans contenaient des molécules d’ADN qui ont permis la détection de Plantes, de Champignons, d’algues et de protistes (Willerslev et coll. 1999).
Détection d’espèces
Une des grandes forces de la méthode de l’ADN environnemental est son potentiel dans les stratégies de conservation appliquée. En effet, elle présente les avantages d’être rapide, peu coûteuse et précise pour détecter spécifiquement des individus, des espèces ou des taxons dans des milieux ouverts. Les objectifs peuvent être complètement différents tout en se basant sur la détection d’espèces rares ou dont la densité est faible. Dans le cas d’une détection d’espèces particulières, le choix se fait au niveau des amorces qui sont utilisées pour amplifier les séquences d’ADN. Ainsi, cela permet une détection précoce d’espèces potentiellement invasives (durant leurs premières étapes d’invasion), ou de pathogènes, et cela à n’importe quel niveau de développement ou quelle que soit la saison. En détectant le plus tôt possible des espèces invasives, les stratégies de confinement et de gestion de ces individus peuvent être adaptées pour augmenter leur efficacité. À titre d’exemple, l’agence américaine de gestion des poissons et de la vie sauvage utilise cette méthode pour suivre l’invasion de la carpe asiatique dans le Midwest (Taberlet et coll. 2012). D’autre part, l’ADN environnemental peut être utilisé dans la détection d’espèces en danger. En effet, par définition, ce sont des espèces avec une faible densité d’individus, qui sont donc difficiles à détecter. Ainsi en détectant leur ADN depuis l’environnement, on peut directement estimer leur présence. Comme autre exemple, on peut également retenir l’utilisation de sangsues pour détecter la présence d’un cervidé très rare du Vietnam, le Muntjac du Truong Son (Schnell et coll. 2012).
Un autre objectif de l’ADN environnemental est de prospecter l’environnement pour détecter de nouvelles espèces. Dans ce cas aussi, le choix des amorces est fondamental : en sélectionnant des amorces universelles pour l’amplification de l’ADN, on peut en théorie détecter indirectement toutes les espèces du milieu. Une étude ayant pour but d’estimer la biodiversité virale dans les océans a permis de détecter environ 33 000 séquences virales dont plus de 65 % étaient inconnues ! (Coutinho et coll. 2017)
Estimation d’abondances
Pour aller plus loin que la simple identification ou détection d’espèces, l’ADN environnemental est également utilisé quantitativement pour estimer des abondances relatives d’individus dans des systèmes naturels (Jerde et coll. 2011, Minamoto et coll. 2012). Cette méthode repose sur un principe de corrélation positive entre la densité d’ADN libre dans l’environnement et la densité d’individus y vivant. Une étude estimant des densités de carpes en milieu expérimental contrôlé a démontré l’existence de cette corrélation (Jerde et coll. 2011). Cependant, cette méthode encore peu précise doit faire face à plusieurs problèmes en milieux naturels : la persistance et la qualité de l’ADN dans le milieu est variable selon son origine (cellules mortes, fèces, sédiments…) et les conditions du milieu, et le nombre de copies d’ADN amplifiées doit être corrélé avec la densité des individus. L’estimation d’abondance d’individus en utilisant leur ADN relâché dans le milieu s’avère donc être un outil potentiellement très utile, notamment en conservation pour estimer des populations d’espèces en danger, mais qui reste peu précis.
Références
- Acinas, S. G., R. Sarma-Rupavtarm, V. Klepac-Ceraj, and M. F. Polz. 2005. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71: 8966–8969.
- Andersen, K., K. L. Bird, M. Rasmussen, J. Haile, H. Breuning‐Madsen, K. H. Kjær, L. Orlando, M. T. P. Gilbert, and E. Willerslev. 2012. Meta-barcoding of ‘dirt’ DNA from soil reflects vertebrate biodiversity. Mol. Ecol. 21: 1966–1979.
- Bienert, F., S. De Danieli, C. Miquel, E. Coissac, C. Poillot, J.-J. Brun, and P. Taberlet. 2012. Tracking earthworm communities from soil DNA: TRACKING EARTHWORM COMMUNITIES FROM SOIL DNA. Mol. Ecol. 21: 2017–2030.
- Bohmann, K., A. Evans, M. T. P. Gilbert, G. R. Carvalho, S. Creer, M. Knapp, D. W. Yu, and M. de Bruyn. 2014. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 29: 358–367.
- Cantera, I., K. Cilleros, A. Valentini, A. Cerdan, T. Dejean, A. Iribar, P. Taberlet, R. Vigouroux, and S. Brosse. 2019. Optimizing environmental DNA sampling effort for fish inventories in tropical streams and rivers. Sci. Rep. 9: 3085.
- CBOL Plant Working Group. 2009. A DNA barcode for land plants. Proc. Natl. Acad. Sci. U. S. A. 106: 12794–12797.
- Coutinho, F. H., C. B. Silveira, G. B. Gregoracci, C. C. Thompson, R. A. Edwards, C. P. D. Brussaard, B. E. Dutilh, and F. L. Thompson. 2017. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 8: 1–12.
- Deagle, B. E., J. P. Eveson, and S. N. Jarman. 2006. Quantification of damage in DNA recovered from highly degraded samples – a case study on DNA in faeces. Front. Zool. 3: 11.
- Dell’Anno, A., and R. Danovaro. 2005. Extracellular DNA Plays a Key Role in Deep-Sea Ecosystem Functioning. Science. 309: 2179–2179.
- Hebert, P. D. N., A. Cywinska, S. L. Ball, and J. R. deWaard. 2003. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270: 313–321.
- Jerde, C. L., A. R. Mahon, W. L. Chadderton, and D. M. Lodge. 2011. “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 4: 150–157.
- Jørgensen, T., J. Haile, P. Möller, A. Andreev, S. Boessenkool, M. Rasmussen, F. Kienast, E. Coissac, P. Taberlet, C. Brochmann, N. H. Bigelow, K. Andersen, L. Orlando, M. T. P. Gilbert, and E. Willerslev. 2012. A comparative study of ancient sedimentary DNA, pollen and macrofossils from permafrost sediments of northern Siberia reveals long-term vegetational stability. Mol. Ecol. 21: 1989–2003.
- Knudsen, B. E., L. Bergmark, P. Munk, O. Lukjancenko, A. Priemé, F. M. Aarestrup, and S. J. Pamp. 2016. Impact of Sample Type and DNA Isolation Procedure on Genomic Inference of Microbiome Composition. mSystems. 1.
- Kunin, V., A. Engelbrektson, H. Ochman, and P. Hugenholtz. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12: 118–123.
- Matheson, C. D., C. Gurney, N. Esau, and R. Lehto. 2010. Assessing PCR Inhibition from Humic Substances. Open Enzyme Inhib. J. 3.
- Minamoto, T., H. Yamanaka, T. Takahara, M. N. Honjo, and Z. Kawabata. 2012. Surveillance of fish species composition using environmental DNA. Limnology. 13: 193–197.
- Nilsson, R. H., M. Ryberg, K. Abarenkov, E. Sjökvist, and E. Kristiansson. 2009. The ITS region as a target for characterization of fungal communities using emerging sequencing technologies. FEMS Microbiol. Lett. 296: 97–101.
- Pompanon, F., B. E. Deagle, W. O. C. Symondson, D. S. Brown, S. N. Jarman, and P. Taberlet. 2012. Who is eating what: diet assessment using next generation sequencing. Mol. Ecol. 21: 1931–1950.
- Schnell, I. B., P. F. Thomsen, N. Wilkinson, M. Rasmussen, L. R. D. Jensen, E. Willerslev, M. F. Bertelsen, and M. T. P. Gilbert. 2012. Screening mammal biodiversity using DNA from leeches. Curr. Biol. 22: R262–R263.
- Shehzad, W., T. Riaz, M. A. Nawaz, C. Miquel, C. Poillot, S. A. Shah, F. Pompanon, E. Coissac, and P. Taberlet. 2012. Carnivore diet analysis based on next-generation sequencing: application to the leopard cat (Prionailurus bengalensis) in Pakistan. Mol. Ecol. 21: 1951–1965.
- Shokralla, S., J. L. Spall, J. F. Gibson, and M. Hajibabaei. 2012. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 21: 1794–1805.
- Taberlet, P., E. Coissac, M. Hajibabaei, and L. H. Rieseberg. 2012. Environmental DNA. Mol. Ecol. 21: 1789–1793.
- Watson, D. L., E. S. Harvey, M. J. Anderson, and G. A. Kendrick. 2005. A comparison of temperate reef fish assemblages recorded by three underwater stereo-video techniques. Mar. Biol. 148: 415–425.
- Willerslev, E., A. J. Hansen, J. Binladen, T. B. Brand, M. T. P. Gilbert, B. Shapiro, M. Bunce, C. Wiuf, D. A. Gilichinsky, and A. Cooper. 2003. Diverse plant and animal genetic records from Holocene and Pleistocene sediments. Science. 300: 791–795.
- Willerslev, E., A. J. Hansen, B. Christensen, J. P. Steffensen, and P. Arctander. 1999. Diversity of Holocene life forms in fossil glacier ice. Proc. Natl. Acad. Sci. 96: 8017–8021.