La mucoviscidose est actuellement traitée à l’aide de médicaments qui améliorent le fonctionnement de la protéine CFTR mutée.
Les cellules qui tapissent notre système respiratoire doivent réguler les flux d’ions et d’eau à travers leurs membranes, afin que notre couche de mucus protectrice ait la bonne consistance. La protéine CFTR (pour cystic fibrosis transmembrane conductance regulator) joue un rôle clé dans cette régulation. Elle laisse passer les ions chargés négativement, tels que les ions chlorures et bicarbonates, et s’ouvre et se ferme selon les besoins pour contrôler l’environnement des voies respiratoires. Lorsque cette fonction est bloquée, les conséquences sont graves. La mucoviscidose est une maladie génétique causée par un gène CFTR défectueux. Le mauvais fonctionnement du canal CFTR entraîne un épaississement du mucus, ce qui provoque une obstruction des voies respiratoires qui peut s’avérer mortelle ainsi que des infections pulmonaires chroniques.
Ouvrir et fermer
La protéine CFTR est similaire à de nombreux transporteurs membranaires de surface, tels que les transporteurs bactériens de résistance multidrogue et notre glycoprotéine P. Ces transporteurs ont une structure caractéristique : une région traversant la membrane forme le pore, tandis que deux domaines liant l’ATP, situé du côté intracellulaire, contrôlent son ouverture et sa fermeture 1. Contrairement à ces autres transporteurs, CFTR comprend également une longue boucle désordonnée qui est phosphorylée par la PKA (protéine kinase dépendante de l’AMPc), ce qui contrôle son activité. La protéine CFTR est également unique parmi ces transporteurs car elle agit comme un canal ionique, permettant aux ions de passer librement lorsqu’elle s’ouvre. Les autres transporteurs similaires font passer des molécules une par une à travers la membrane.
Améliorer la fonction
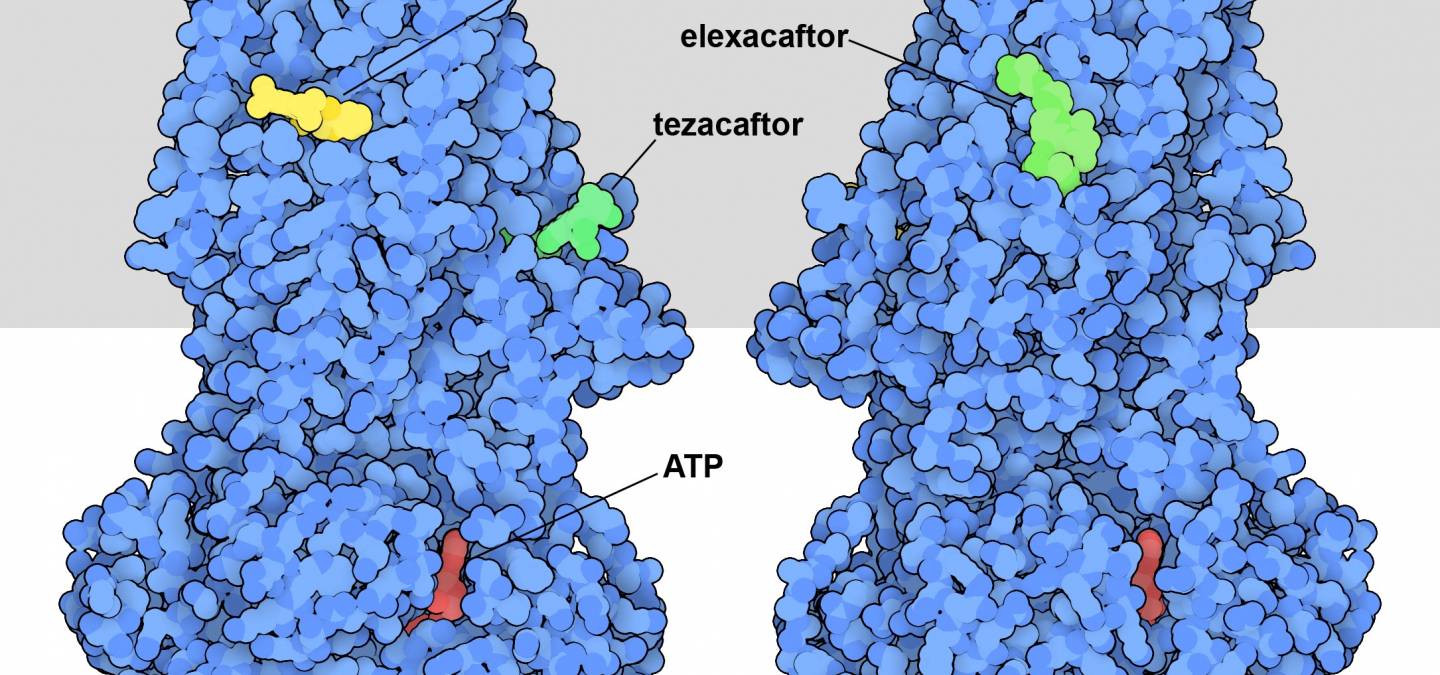
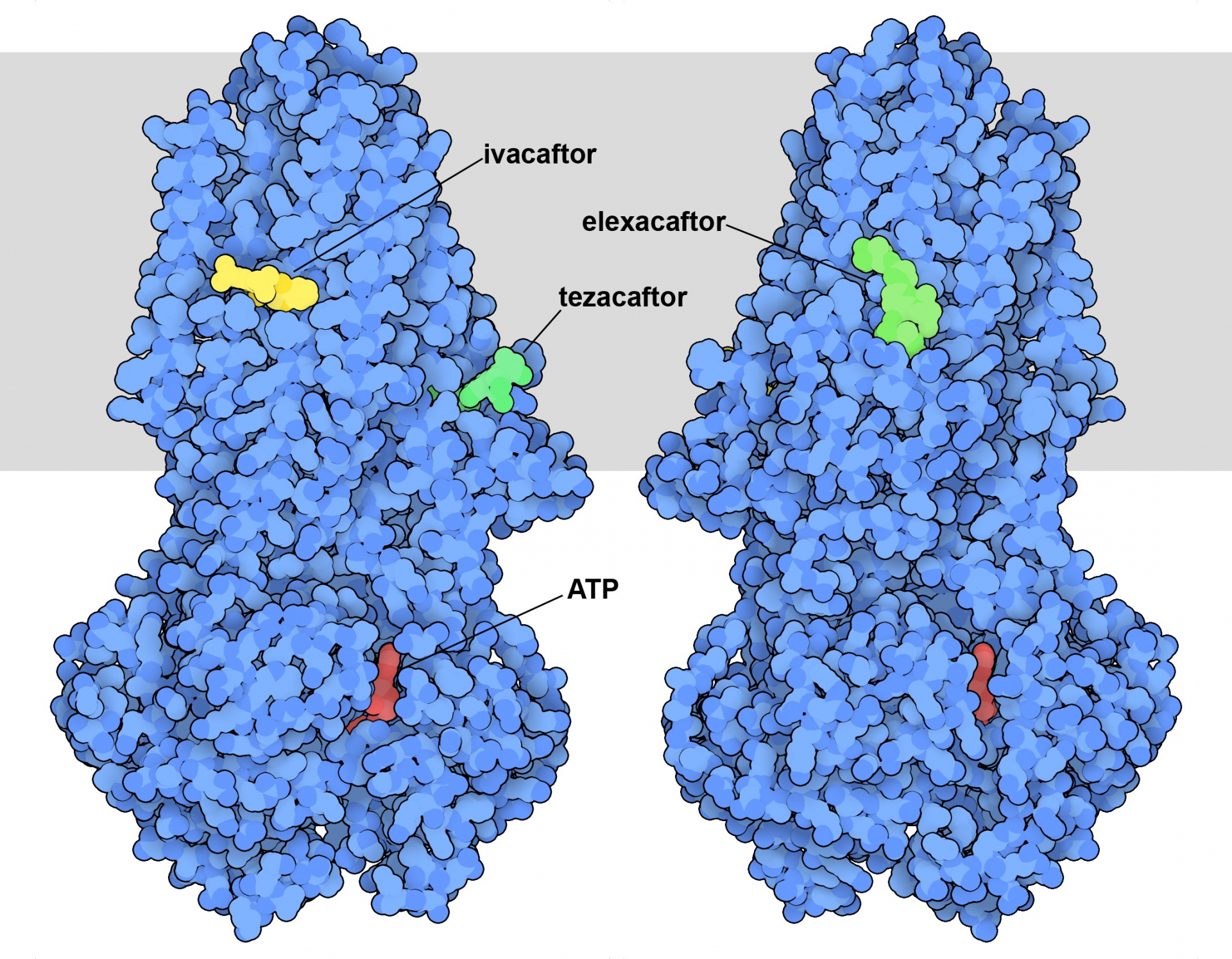
La connaissance de la structure et de la fonction de la protéine CFTR a permis la découverte de médicaments pour traiter la mucoviscidose. L’entrée 8eiq de la banque de données sur les protéines (PDB) montre trois médicaments qui sont souvent utilisés ensemble pour traiter les personnes porteuses de la variante la plus courante de la protéine CFTR à l’origine de la maladie (Figure 1) 1. Ils agissent d’une manière différente de la plupart des médicaments : au lieu de bloquer l’action de CFTR, ils améliorent sa fonction. La suppression d’un seul acide aminé dans la protéine CFTR, la phénylalanine 508, déstabilise la protéine. Cela entraîne de nombreux problèmes. Tout d’abord, lorsqu’elle est synthétisée dans les cellules pulmonaires, celles-ci perçoivent qu’il y a une anomalie et détruisent la majeure partie de la protéine. Ces cellules ont donc moins de protéines CFTR mutées à leur surface. Deuxièmement, la protéine déstabilisée n’est pas aussi efficace en tant que canal ionique. Les médicaments corrigent ces deux problèmes. L’ivacaftor se lie à la région formant le pore et améliore la stabilité et l’efficacité de celui-ci. L’elexacaftor et le tezacaftor, quant à eux, contribuent à la stabilité de la protéine au cours de sa biosynthèse, garantissant ainsi la présence d’une plus grande quantité de la protéine à la surface de la cellule.
Nouvelles cibles thérapeutiques
CFTR n’est pas la seule protéine qui gère l’équilibre des fluides dans les voies respiratoires pulmonaires. D’autres protéines sont également étudiées dans l’espoir de trouver d’autres moyens de traiter la mucoviscidose. Le canal sodique épithélial (ENaC, entrée PDB 6bqn) en est un exemple 1. Comme son nom l’indique, il gère le flux d’ions sodium à travers les membranes plasmiques.
CFTR active et inactive
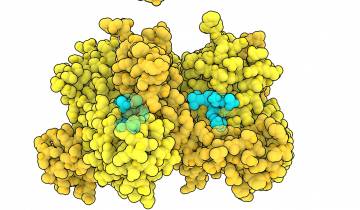
Comme le montrent les entrées PDB 5uak et 6msm, la protéine CFTR subit un important changement de conformation lorsqu’elle devient active. Les deux domaines de liaison à l’ATP sont séparés dans la forme inactive, le canal ionique est alors fermé 1. Lorsque l’ATP (rouge) se lie, les deux domaines de liaison à l’ATP se rejoignent, ce qui ouvre le canal 2. La phénylalanine 508 est représentée en turquoise vif. Remarquez qu’elle stabilise la jonction entre la partie transmembranaire et l’un des domaines de liaison à l’ATP.

Pour aller plus loin
La protéine CFTR comprend un domaine régulateur qui est largement désordonné et qui n’est donc pas représenté dans les structures atomiques. Cependant, le site PDB-101 propose un modèle structurel calculé (et non observé, NdT) de ce domaine régulateur, qui donne une idée de sa taille.
Ce texte correspond à la traduction par Cédric Bordi de l’article Molecule of the Month : CFTR and Cystic Fibrosis 1 écrit par David S. Goodsell et paru en mai 2024 sur le site PDB-101, le portail éducatif de la base de données sur les protéines (PDB).
Crédits

Cet article est publié en partenariat avec le site PDB-101, le portail éducatif de la base de données sur les protéines (PDB).