

La trisomie 21 peut avoir de nombreuses origines. Cet article présente les différents mécanismes pouvant mener à cette maladie (anomalies lors de la méiose, translocations chromosomiques...).
Cet article est une version révisée, grâce aux apports de Rodolphe Dard, de celle publiée en novembre 2003 par Gilles Furelaud.
La trisomie 21 est l’anomalie chromosomique la plus fréquente dans l’espèce humaine. Si ses grandes caractéristiques sont en général connues, ses origines précises peuvent être plus diversifiées que l’on est souvent tenté de le penser.
Qu’est-ce que la trisomie 21 ?
Une cellule humaine diploïde possède 23 paires de chromosomes, soit 46 chromosomes. Chaque paire étant identifiée par un numéro, les paires 1 à 22 sont les autosomes tandis que la 23ᵉ paire correspond aux chromosomes sexuels X et Y, appelés gonosomes. Le génome humain est donc porté par 24 chromosomes différents (1 à 22 + X + Y), abstraction faite de l’ADN mitochondrial.
Les chromosomes, correspondant à de l’ADN condensé autour de structures protéiques, n’adoptent leur forme classique de bâtonnets que lors de la mitose ou de la méiose. Durant le reste du cycle cellulaire, l’ADN est débobiné, et présent sous forme de filament, non visualisable au microscope optique. Le caryotype, est l’examen des chromosomes au microscope. Il est possible d’observer les 46 chromosomes sur toute cellule du corps humain en division spontanée, ou après induction de celle-ci. Il est à noter que les cellules sexuelles sont haploïdes et ne possèdent donc que 23 chromosomes.
On observe chez certaines personnes des aberrations chromosomiques pouvant se traduire soit par un nombre anormal de chromosomes (aneuploïdie), soit par des chromosomes remaniés (anomalie de structure), les deux pouvant être liés (cf infra). Les aneuploïdies peuvent concerner les 22 paires d’autosomes et les chromosomes sexuels (45 X ou syndrome de Turner, 47 XXY ou syndrome de Klinefelter, avec alors des conséquences importantes mais non létales – lire à ce sujet La mise en place de l’appareil génital chez l’être humain – ou des variantes sans conséquence pathologique : 47 XXX, 47 XYY).
Lorsque l’aneuploïdie concerne une paire de chromosomes, il peut s’agir :
- d’une monosomie (un seul chromosome) ; par exemple le syndrome de Turner 45, X
- d’une trisomie (trois chromosomes) ; par exemple la trisomie 18 : 47, XY + 18 ou 47, XX + 18
- d’une tétrasomie (quatre chromosomes) ; par exemple le syndrome de Pallister-Killian avec quatre copies du bras court (12p) du chromosome 12.
Lorsque l’aneuploïdie concerne l’ensemble du lot chromosomique on parle de triploïdie (69 chromosomes), tétraploïdie (92 chromosomes), etc.
Les aneuploïdies sont pour la majorité létale à un stade très précoce de développement embryonnaire. Elles sont par ailleurs fréquentes dans l’espèce humaine, puisqu’on estime qu’environ 30 % à 50 % des grossesses passeraient inaperçues par une fausse couche très précoce dans les deux premières semaines, suite à une anomalie du nombre de chromosomes (triploïdie surtout).
Toutefois certaines aneuploïdies sont compatibles avec une grossesse évolutive, sans pour autant être viables. D’autres peuvent exceptionnellement être menées à terme, mais avec un pronostic très sombre et un décès très souvent avant le premier mois de vie (trisomie18, notée T18, et trisomie 13, notée T13). Enfin certaines sont couramment compatibles avec une grossesse normale et une espérance de vie importante (trisomie 21, T21 ; syndrome de Turner…).
Il faut retenir que les principales manifestations de ces aneuploïdies sont des anomalies malformatives souvent multiples (malformation cardiaque, digestive, cérébrale…), et pour celles « viables », un retard de développement neurologique.
Les trisomies 13, 18 et 21 sont les seules viables dans l’espèce humaine. Elles sont relativement courantes (1/500 à la naissance pour la T21, 1/10 000 pour la T18 et 1/20 000 pour la T13).
La trisomie 21, ou syndrome de Down pour les anglo-saxons, est donc à la fois l’anomalie chromosomique la plus courante à la naissance, et une pathologie dont l’espérance de vie est importante et grandissante. En effet l’espérance de vie, en France, en 1960, des patients porteurs d’une trisomie 21 était de 9 ans, alors qu’aujourd’hui elle est de plus de 60 ans pour la moitié de ces patients. Cette augmentation spectaculaire de l’espérance de vie est multifactorielle :
-
Les avancées de la médecine ont permis de mieux prendre en charge ces enfants aux comorbidités multiples. Les progrès chirurgicaux notamment ont permis d’améliorer la survie des patients porteurs d’une cardiopathie congénitale ou d’une malformation digestive qui concernent tout de même plus de la moitié des patients à la naissance (en condition « sauvage », c'est-à-dire en l'absence de dépistage prénatal). La prise en charge de l’hypothyroïdie et de la sensibilité aux infections a également été essentielle. Malgré tout, on sait que la présence d’une cardiopathie ou d’une malformation digestive demeure de mauvais pronostic avec une mortalité de 20 à 30 % dans les cinq premières années de vie.
-
Le dépistage prénatal aboutit à la sélection des embryons les moins mal formés et qui ont donc une meilleure espérance de vie.
-
La prise en charge sociale et médicale a grandement évolué, passant d’enfants non médicalisés et isolés, à une prise en charge active (la trisomie 21 a longtemps été une contre-indication à de nombreux actes médicaux), médicale, paramédicale et sociale. Malheureusement, des inégalités demeurent, avec une espérance de vie en relation directe avec le niveau social et l’isolement géographique.
La trisomie 21 se caractérise, comme son nom l’indique, par la présence de trois chromosomes 21. Les personnes porteuses de cette anomalie présentent diverses caractéristiques : nuque large, visage de forme spécifique (d’où dérive le nom de « mongolisme » autrefois donné à la maladie), malformations viscérales (cardiopathie, malformation digestive), problèmes métaboliques et retard mental plus ou moins important. Concernant la fertilité des hommes et femmes porteuses d’une T21, la littérature ancienne fait état d’une stérilité masculine, qui n’est absolument pas documentée et semble infirmée. Les femmes ont une fertilité normale. Bien sûr, le risque de récidive de trisomie 21 dans ces cas-là est important mais mal documenté.
Toutefois, comme nous allons le voir, la trisomie 21 peut avoir une cause chromosomique plus subtile que la « simple » présence de trois chromosomes 21.
Les différentes origines de la trisomie 21
La présence de trois chromosomes 21
Dans sa forme la plus courante, la trisomie 21 se caractérise par la présence de trois chromosomes 21. En général, l’origine de cette trisomie est une fécondation entre un gamète possédant un chromosome 21 et un gamète possédant deux chromosomes 21. Dans la très grande majorité des cas, l’anomalie est portée par l’ovocyte. En effet, le vieillissement des ovocytes est à l’origine d’anomalies méiotiques favorisant la survenue d’une trisomie. Cet aspect physiopathologique explique que le risque de T21 soit directement lié à l’âge maternel.
Normalement, un gamète possède un seul chromosome 21. La présence de deux chromosomes 21, peut provenir d’une non-disjonction des chromosomes homologues (lors de la première division de méiose) ou des chromatides sœurs (lors de la seconde division de méiose). La trisomie 21 est alors dite libre et homogène. Libre, car il y a bien trois chromosomes 21, non liés à un autre chromosome, donc une formule à 47 chromosomes. Homogène, car l’ensemble des cellules du zygote porte la trisomie. Ces T21 libres et homogènes sont de novo, accidentelles, avec un risque de récidive pour de futures grossesses estimé à 1 % de manière empirique (même aneuploïdie ou autre anomalie chromosomique, ainsi une T21 peut récidiver en T13 par exemple).
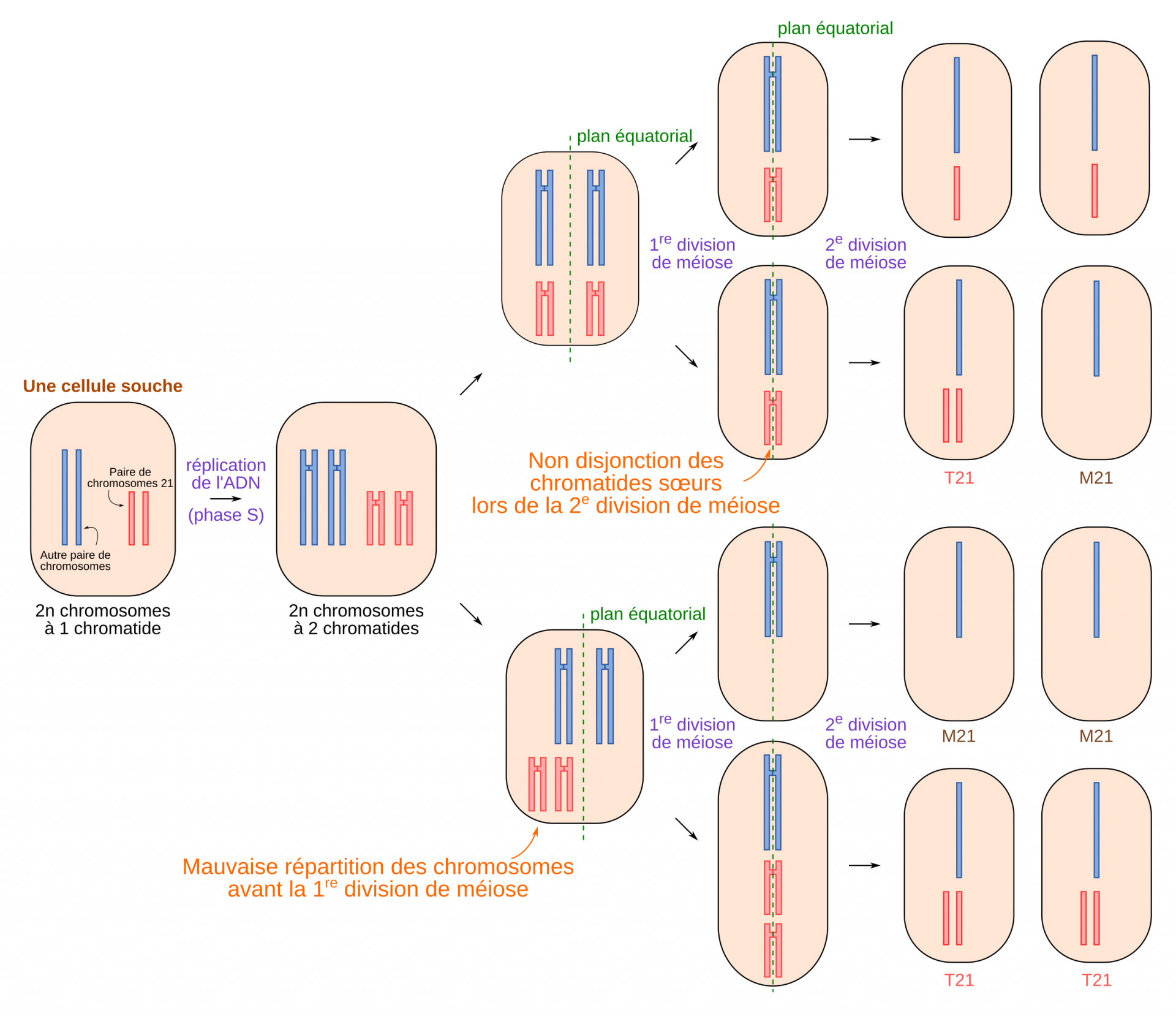
Des gamètes contenant deux chromosomes 21 peuvent être formés à cause d’anomalies lors de la première ou lors de la seconde division de méiose. Si un tel gamète féconde un gamète possédant un chromosome 21, l’embryon résultant possédera trois chromosomes 21. De telles méioses produisent également des gamètes sans chromosome 21. Cependant des embryons issus de ces gamètes ne sont pas viables.
Une trisomie 21 libre peut avoir d’autres causes :
- Il est possible que l’un des deux parents soit déjà trisomique 21 : celui-ci transmet alors un ou deux chromosomes 21 à ses enfants.
- Il est possible que la non-disjonction de chromosomes ait lieu lors d’une division de mitose :
- chez l’un des parents (non trisomique), lors de la division d’une cellule précurseure des gamètes ;
- chez la personne trisomique, au tout début de son développement embryonnaire. Dans ce dernier cas, on assiste à une trisomie 21 mosaïque : seule une partie des cellules de l’individu sont trisomiques. Le phénotype des individus est variable, en fonction des organes touchés.
Le phénotype des trisomiques 21 s’explique par la présence en triple exemplaire de certains gènes portés par le bras long du chromosome 21. Par conséquent, d’autres anomalies chromosomiques que celles décrites jusqu’ici peuvent conduire à ce phénotype : il suffit pour cela que le bras long du chromosome 21 soit présent en triple exemplaire. C’est ce qui arrive en cas de translocation d’un chromosome 21 sur un autre chromosome. On observe alors une trisomie 21 non libre, avec une formule à 46 chromosomes. La translocation à l’origine de la T21 peut être réciproque ou robertsonienne.
Les translocations réciproques
(A) Une translocation réciproque correspond à un échange de matériel entre deux chromosomes. (B) Si un des gamètes contient un chromosome dérivé de translocation, l’embryon possédera donc trois morceaux de chromosomes 21 et présentera donc le phénotype trisomique 21.
Dans le cas de la translocation réciproque (Figure 2), il y a un échange de matériel entre deux chromosomes. Cela peut concerner n’importe quel autosome ou gonosome. Ainsi, en cas de translocation réciproque 7;21, un fragment terminal de chromosome 7 est échangé contre un fragment terminal de chromosome 21. On se retrouve donc avec un chromosome 7 tronqué et ayant hérité d’un morceau de 21, et un chromosome 21 tronqué ayant hérité d’un morceau de chromosome 7. Ces chromosomes sont appelés « dérivés de translocation ». Bien sûr, ils sont en paires avec, respectivement, un chromosome 7 normal, et un chromosome 21 normal. L’individu porteur de cette translocation réciproque, porte 46 chromosomes. Il est en bonne santé, car tout le matériel chromosomique est présent en quantité normale, mais « mal rangé », ce qui n’occasionne pas de phénotype particulier. On parle de translocation équilibrée.
En revanche, en cas de grossesse il y a un risque important de transmission d’une anomalie chromosomique déséquilibrée. L’individu peut en effet transmettre son chromosome 7 anormal et son chromosome 21 normal. Dans ce cas on a donc un zygote qui porte trois fois le même fragment de chromosome 21 (celui de chacun des deux chromosomes 21 et celui présent sur le chromosome 7). Cet individu a une trisomie 21 partielle, mais également une monosomie partielle du chromosome 7 (cf figure 3). Dans ces circonstances, le zygote n’aura pas de trisomie 21 partielle si et seulement si son parent lui a transmis ses chromosomes 7 et 21 normaux ensemble, ou ses chromosomes 7 et 21 remaniés ensemble. La genèse du déséquilibre est liée à la formation d’appariements non pas deux à deux des chromosomes homologues lors de la méiose, mais 4 à 4 (figure tétravalentes), avec donc 16 possibilités de disjonction. On comprend également que de nombreuses possibilités sont très déséquilibrées et donneront des fausses couches précoces. Ainsi ce type d’anomalie est en général héréditaire, transmis sur plusieurs générations, avec des problèmes de fertilité et des fausses couches à répétition.
Ces translocations peuvent être héritées d’un des deux parents ou bien se faire de novo, c’est-à-dire apparaître lors de la méiose chez l’un des deux parents, ou lors des premières mitoses de l’embryon.
Les translocations robertsoniennes
(A) Une translocation robertsonienne correspond à la fusion de deux chromosomes acrocentriques. (B) Si un des gamètes contient un chromosome dérivé de translocation, l’embryon possédera donc trois morceaux de chromosomes 21 et présentera donc le phénotype trisomique 21.
Un autre type de translocation existe. Il s’agit des translocations dites robertsoniennes. Dans ce cas on observe la fusion complète de deux chromosomes différents ; par exemple d’un chromosome 21 complet avec un chromosome 14 complet. L’individu porteur d’une telle translocation équilibrée, a donc 45 chromosomes et est en bonne santé (par exemple : 1 chromosome 21 + 1 chromosome 14 + 1 chromosome 14;21, soit 3 chromosomes au lieu de 4). Tous les chromosomes ne peuvent pas donner de translocation robertsonienne. Seuls les chromosomes acrocentriques sont concernés. Il y en a cinq dans l’espèce humaine, les chromosomes 13, 14, 15, 21 et 22 (groupes D et G du caryotype). La particularité de ces chromosomes est d’avoir un centromère très « haut », avec un bras court (bras p), quasi inexistant, et porteur uniquement d’hétérochromatine avec des séquences répétées (microsatellites) et dépourvues de gènes. Les bras courts ayant peu d’importance et étant relativement similaires d’un chromosome acrocentrique à un autre, ils peuvent être perdus et provoquer la fusion de deux chromosomes acrocentriques. Ainsi on peut observer une translocation 13;21 ou même 21;21.
Seuls les chromosomes acrocentriques peuvent être à l’origine de translocations robertsoniennes. Il s’agit des chromosomes du groupe D (13, 14 et 15) et du groupe G (21 et 22).
À l’image de la transmission des translocations réciproques, lors de la méiose se forment des figures trivalentes, pouvant entraîner des déséquilibres lors de la disjonction et donc l’apparition d’une trisomie 13, 18 ou 21 selon la translocation à l’origine de l’anomalie.
Les risques de récidive
Un point intéressant est que le risque de récidive, c’est-à-dire la probabilité d’avoir un enfant de phénotype type trisomie 21, est différent selon les translocations (on parle de récidive car ces analyses chromosomiques sont réalisées en général suite à la naissance d’un premier enfant atteint de trisomie 21). Ce risque est à calculer au cas par cas pour les translocations réciproques, selon les points de cassures chromosomiques ; il est bien connu pour les translocations robertsoniennes. Il est également intéressant de noter que ce risque est différent si l’anomalie est de transmission maternelle ou paternelle.
Tableau 1 : Risque pour un individu possédant une translocation robertsonienne d’avoir un enfant atteint de trisomie 21, selon le type de translocation
| Père | Mère | |
|---|---|---|
| t(Dq ; 21q) | 5 % | 15 % |
| t(22q ; 21q) | 5 % | 15 % |
| t(21q ; 21q) | 100 % | 100 % |
| Comment lire ces translocations : t(a ; b) = translocation robertsonienne entre le chromosome « a » et le « b » Spécification des chromosomes : D = chromosome 13, 14 ou 15 q = bras long du chromosome |
||
On constate donc qu’un parent porteur d’une translocation du chromosome 21 sur lui-même (translocation dite robertsonienne) est assuré d’avoir un enfant atteint. En effet les deux bras longs des chromosomes 21 sont désormais portés par un même « chromosome ». Ses gamètes sont donc assurés d’être soit porteurs de ce « chromosome », soit sans chromosome 21 du tout. Après fécondation, on obtient donc un embryon qui est soit trisomique 21 (trois bras longs 21), soit monosomique 21. Or la monosomie 21 est létale. Seuls les trisomiques 21 survivent.
Documents à télécharger
Animation Flash présentant les différentes origines possibles de la trisomie 21
Quelques chiffres
Toutes ces origines possibles ne contribuent pas dans les mêmes proportions aux personnes atteintes de trisomie 21.
| Trisomie 21 libres et homogènes (trois chromosomes 21 dans toutes les cellules de l’organisme) | 92,5 % |
| Trisomie 21 mosaïque (seules certaines cellules présentent trois chromosomes 21, les autres cellules en ont deux) | 2,5 % |
| Translocations (présence de trois bras longs ou fragments de bras longs du chromosome 21) | 4 % |
| Autres cas | 1 % |
Il est possible de préciser ces origines. Par exemple, dans le cas d’une trisomie 21, la non-disjonction des chromosomes ou des chromatides peut avoir eu lieu lors de la première division de méiose (méiose I) ou lors de la deuxième (méiose II).
| Père | Mère | |
| Méiose I | 5 % | 70 % |
| Méiose II | 5 % | 20 % |
| Total | 10 % | 90 % |
Conclusion
La trisomie 21 est la principale altération du caryotype à la naissance. Toutefois, sous le terme très général de trisomie 21, ou syndrome de Down, on regroupe en fait une multitude de cas différents, aussi bien par l’explication du phénotype (trisomie libre et homogène, translocation, etc.) que par la gravité du phénotype. En effet, de multiples graduations sont observées dans l’atteinte des individus : ceci est dû à une modulation par l’ensemble du génome, à la présence d’individus mosaïques, à l’importance du fragment de chromosome concerné dans les cas de translocations, etc.
Si la trisomie 21 est si importante en termes de nombre d’individus atteints, c’est parce que c’est l’une des rares anomalies chromosomiques viables. Un dépistage prénatal (échographies, analyses biochimiques, voire caryotype, etc.) est ainsi mené lors du suivi d’une grossesse. L’importance de ce dépistage (en particulier le choix de réaliser un caryotype) dépend de certains signaux d’alerte : âge maternel, antécédents, observations (clarté nucale et autres mesures à l’échographie) et analyses (dosages sanguins maternels en particulier).



