

L'expédition Tara Océans, menée entre 2009 et 2013, avait notamment pour objectif de mieux caractériser le plancton. Les études génétiques ont permis d'identifier les espèces présentes en différentes régions du globe, mais aussi des montrer que les océans renferment de nombreuses espèces inconnues. La détermination des gènes présents dans l'océan permet également de faire des déductions sur les caractéristiques, notamment écologiques, des espèces ainsi mises en évidence.
L’importance du plancton
L’océan représente le plus grand écosystème continu sur Terre, et 98 % de sa biomasse est composée d’organismes qui sont invisibles à l’œil nu : les micro-organismes marins, dont nombre d’entre eux vivent comme « plancton ». Le terme « plancton » vient du grec planktos, ou « errant » ; il désigne les organismes qui vivent dans la colonne d’eau et sont incapables de nager contre le courant. Le plancton marin est ainsi composé de bactéries, de protistes (des eucaryotes unicellulaires), de virus, d’archées, mais aussi des stades larvaires d’organismes plus gros, comme les larves de poissons ou de crustacés.
Alors que les membres du plancton de l’ordre du millimètre ont été étudiés depuis plus d’un siècle, ce n’est que depuis les années 1970 que l’abondance et la diversité des microbes et virus ont été mises au jour. Il a été montré depuis qu’un litre d’eau de mer peut contenir jusqu’à 109 bactéries. Dans les années 1990, les premières techniques d’évaluation de la diversité bactérienne marine indépendantes de mise en culture – notamment génétique – montrent que les bactéries sont diverses, et que la majorité des groupes marins étaient précédemment inconnus. Simultanément, la découverte des virus dans l’océan, atteignant presque 1010 particules/L, rajoute une nouvelle couche de complexité dans notre compréhension de ce qui génère, et maintient la diversité microbienne. La diversité des protistes – eucaryotes unicellulaires – est révélée dans les années 2000, à travers l’usage de la génétique, et rend notre connaissance de la diversité planctonique assez récente.

Les protistes forment un groupe polyphylétique correspondant aux eucaryotes unicellulaires. Les taxons représentés sont soit formés uniquement d'unicellulaires, soit d'unicellulaires et de pluricellulaires (par exemple, Chloroplastidés et Eumycètes). Seuls les Métazoaires ne contiennent que des êtres vivants pluricellulaires. Autrement dit, à l'exception du groupe des Métazoaires, on trouve des protistes dans tous les groupes d'Eucaryotes notés ici. D’après Lecointre et Le Guyader, 2016.
L’exploration de la distribution globale des communautés et diversités de microbes marins devient quantitative grâce au séquençage à haut débit disponible au milieu des années 2000, et ouvre la voie pour des campagnes d’échantillonnage à grande échelle spatiale. En effet, la distribution du plancton dépend fortement de facteurs abiotiques, comme la lumière, les nutriments, la turbulence, la température, la salinité ou le pH, et de facteurs biotiques, comme la présence d’autres organismes tels des prédateurs ou des symbiontes. Même si l’abondance locale du plancton varie de façon horizontale, verticale et saisonnière, les organismes planctoniques sont présents partout dans les océans.
L’importance du plancton à l’échelle planétaire est multiple. Il est à la base des chaînes alimentaires et représente 50% de la production annuelle de dioxygène sur Terre (Field, 1998). Le métabolisme du plancton joue un rôle majeur dans les grands cycles biogéochimiques du carbone, de l’oxygène, de l’azote, du phosphore et du soufre.
L’expédition Tara Océans
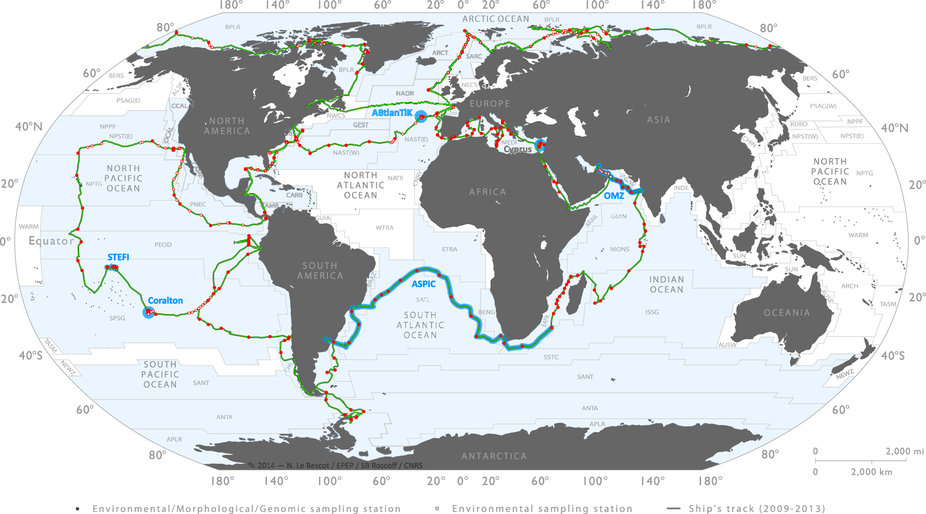
L’expédition Tara Océans (2009-2013) a eu pour objectif de découvrir la grande variété d’organismes planctoniques (des virus aux larves de poissons) de l’océan de surface (entre 0 et 200 m) et mésopélagique (200 à 1000 m) à l’échelle planétaire. Au total, 40 000 échantillons d’eau de mer et de plancton ont été prélevés dans 210 stations réparties dans 20 provinces biogéographiques. De nombreuses questions ont animé cette expédition : quelle est la vraie nature de la diversité planctonique dans nos océans ? Quels sont les organismes qui portent les fonctions les plus importantes ? Quel est l’effet des paramètres environnementaux et des interactions biotiques sur l’écosystème océanique ?
Tara expéditions
Le site Tara expéditions propose de nombreux outils pédagogiques : kits de données, activités et expériences à réaliser en classe. Vous pourrez notamment y retrouver un ensemble de données sur l'analyse génétique des échantillons de plancton.

Afin de répondre à ces questions, Tara a regroupé plus de 100 scientifiques à travers le monde et procédé à un échantillonnage strictement identique pendant plus de trois ans sur la goélette de 36 mètres.
Le programme d’échantillonnage standard a été conçu pour étudier une grande variété d’écosystèmes marins : remontées d’eau (upwellings), points chauds de biodiversité, zones de bas pH ou pauvres en dioxygène… Un total de 210 stations a été défini sur lesquelles une caractérisation environnementale plus précise a été conduite, afin de contextualiser les prélèvements morphologiques et génétiques du plancton. Pour chaque station, les prélèvements d’eau ont été filtrés puis soumis à différentes analyses génétiques :
- séquençage de l’ADNr 18S (metabarcoding) ;
- métatranscriptomique : détermination de l’ensemble des transcriptomes présents, c’est-à-dire de l’ensemble des ARN produits par les différents organismes ;
- métagénomique : caractérisation de l’ensemble des génomes.
Des échantillons d’eau préservés grâce à du paraformaldéhyde sont utilisés pour la microscopie à haute résolution, tandis que d’autres sont fixés dans de l’éthanol ou du lugol pour permettre d’examiner les populations ultérieurement, pour des projets auxquels nous n’aurions pas encore pensé.
Tara et la génétique
Le séquençage ADN à haut débit ouvre la porte de l’exploration de la diversité génétique microbienne d’échantillons environnementaux, à la fois qualitative et quantitative. Elle permet de comprendre qui est là, qui fait quoi, et quel est le répertoire de gènes présent dans l’océan.
Qui est là ?
Barcoding
La phylogénie des micro-organismes a longtemps été fondée sur des caractères morphologiques et biochimiques. Récemment, les marqueurs moléculaires (barcodes) ont été utilisés pour reconstruire l’histoire évolutive des organismes vivants, basé sur l’idée – simplifiée – que plus deux organismes sont distants évolutivement, plus la différence entre leur séquence génomique sera grande. Stricto sensu, un code-barre ADN (barcode) est une courte séquence (typiquement 100 à 400 paires de bases) correspondant à une portion standard du génome (par exemple ADN ribosomal 18S), qui peut être utilisée pour identifier les espèces, comme un code-barre utilisé au supermarché qui permet de faire la relation entre le produit (la séquence) et son prix (l’identification de l’espèce). Cette séquence est choisie sur la base de critères très précis (Valentini et al., 2009) : la variabilité intra-espèce doit être faible (cette séquence doit être quasi identique chez tous les organismes de la même espèce) et en même temps la variabilité inter-espèces doit être grande (qu’on puisse différencier deux espèces différentes sur la base de leur séquence).
Les marqueurs moléculaires ADN les plus utilisés pour la reconstruction phylogénétique sont les gènes codants les ARNr des sous-unités ribosomales, les ADNr. Chez la majorité des eucaryotes, l’ARNr 18S est présent dans la petite sous-unité ribosomale, tandis que la grande sous-unité contient trois molécules d’ARNr (5S, 5,8S, 28S chez les mammifères et 25S chez les plantes). Les gènes codant les ARNr sont souvent regroupés en batterie (cluster), et séparés par des espaceurs internes transcrits (internal transcribed spacers, ITS1 et ITS2) et un espaceur intergénique (IGS, Figure 4).
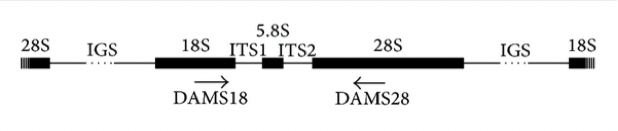
Organisation typique d’une batterie (cluster) d’ADNr chez les eucaryotes. Les gènes 18S, 5,8S et 28S codent les ARN ribosomaux ; ITS1 et ITS2 sont les espaceurs internes transcrits (internal transcribed spacers); Les espaceurs intergéniques (IGS : intergenic spacers) séparent les nombreuses copies de ces gènes.
Metabarcoding
L’impact des code-barres comme outil moléculaire va bien au-delà d’une phylogénie à plus grande résolution des espèces connues : conservation, découvertes d’espèces, et écologie des communautés en bénéficient (Kress et al., 2015). Grâce à l’arrivée du séquençage à haut débit, le barcoding est devenu un outil répandu pour l’écologie des communautés eucaryotes (et procaryotes !) à travers une méthode connue sous le nom de metabarcoding, « meta » exprimant l’idée que le barcoding est réalisé non pas sur une seule espèce mais sur un ensemble d’espèces provenant d’échantillons environnementaux.
Plus formellement, « le metabarcoding ADN fait référence à l’identification automatisée de plusieurs espèces provenant d’un même échantillon contenant les organismes entiers, ou d’un échantillon environnemental contenant de l’ADN dégradé (du sol, de l’eau, fèces, etc.) » (Taberlet et al., 2012). Un bon code-barre pour une étude metabarcoding (comme l’ADNr 18S) doit (i) correspondre à une portion de gène quasi identique au sein des individus de la même espèce, mais différent entre espèces (ii) être utilisable pour toutes les espèces considérées dans l’étude, (iii) permettre l’assignation taxonomique et (iv) ce à différents niveaux taxonomiques (Valentini et al., 2009). Après extraction d’ADN de l’échantillon, une PCR avec des amorces dites « universelles » est réalisée, qui permet d’amplifier la portion choisie (par exemple, l’ADNr 18S) de tous les organismes présents.
Comment construire des amorces universelles, c’est-à-dire capables d’amplifier l’ADN de n’importe quel organisme présent dans un échantillon ? L’idée est de partir des séquences d’ADNr 18S déjà connues et de comparer ces séquences (on parle d’alignement) entre divers organismes. Cette démarche a permis d’identifier une région appelée V9, longue de 130 paires de bases dont les extrémités sont très conservées chez tous les organismes connus. Les extrémités de cette séquence peuvent donc servir de point d’ancrage pour les amorces lors de la PCR. Par construction, il est possible que ces amorces ne détectent pas certains micro-organismes absents des bases de données de références, qui auraient une séquence V9 relativement différente de celle des autres Eucaryotes. C’est ainsi que des recherches ont pu montrer que des amorces, que l’on pensait universelles, ne l’étaient pas en réalité.
Le produit PCR final est donc un mélange de toutes les séquences V9 des organismes présents. Chacune des copies est ensuite séquencée : on obtient l’ensemble de la diversité des V9 de notre échantillon. Les séquences présentant un degré de similarité de 97 % ou plus sont regroupées en unités taxonomiques opérationnelles (UTO, ou OTU en anglais pour operational taxonomic unit) - (Blaxter et al., 2005). Il faut ensuite faire correspondre à chaque UTO une espèce. Cette assignation taxonomique se fait en comparant les UTO de l’échantillon avec des bases de données de référence d’organismes en culture. L’idée est de voir de quelle séquence connue l’UTO se rapproche le plus, et dans quelle proportion. Le degré de similarité permettra de donner avec un certain degré de certitude un nom d’espèce, ou de genre, ou de famille. Seul hic, plus de 40 % des UTO déterminées dans le cadre de Tara Océans sont totalement nouvelles et ne correspondent à aucune espèce connue. On sait simplement qu’il s’agit d’Eucaryotes, mais il est quasiment impossible de les placer dans un arbre phylogénétique. Excitant !
Il faut noter que ces technologies de metabarcoding évoluent aussi vite que baisse le prix du séquençage. D’autres marqueurs plus longs, tels le V4 de 380 paires de bases, auparavant trop chers, sont maintenant accessibles. Leur plus grande taille augmente la résolution de la phylogénie. Aujourd’hui, et principalement pour le domaine procaryote, d’autres approches s’appuient sur les séquençages de génomes entiers, ou métagénomique. Au lieu d’utiliser des amorces spécifiques, l’ensemble des génomes dans l’échantillon environnemental est séquencé. La première étape, après extraction de l’ADN, consiste à fragmenter tous les ADN présents dans l’échantillon en morceaux très courts puis à les séquencer ; on parle de shotgun sequencing. Par la suite, les fragments séquencés sont assemblés bioinformatiquement à partir des régions chevauchantes, afin de reconstruire les génomes d’origines. Il est possible d’identifier des portions clefs (les code-barres) a posteriori, appelés « miTags » (Logares et al., 2014), sur la base de leur ressemblance avec d’autres code-barres de la même famille. On identifie ainsi une portion du génome séquencé qui ressemble à l’ADNr 16S (pour les procaryotes) ou 18S (pour les eucaryotes). La démarche est donc différente de celle du metabarcoding où ces régions étaient ciblées a priori par la PCR. L’approche miTags permet ainsi d’identifier les code-barres au sein des génomes, mais aussi les gènes présents.
Qui fait quoi et qui peut faire quoi ?
Au-delà de faire un inventaire des espèces présentes, l’échantillonnage de Tara a aussi eu recours aux techniques de métatranscriptomique. Elles permettent de rendre compte des gènes exprimés dans l’échantillon en séquençant les ARN messagers (ARNm). Pour étudier spécifiquement les transcrits eucaryotes, il est possible de sélectionner uniquement les ARNm polyadénylés (dotés en 3’ d’une queue faite d’une succession d’adénosine, absente chez les procaryotes). Ces ARNm sont ensuite rétrotranscrits et les ADN complémentaires obtenus sont séquencés et comparés à des bases de données de références pour essayer à la fois de donner une annotation fonctionnelle (de quels gènes ces transcrits proviennent-ils ? à quoi peuvent-ils servir ?) et taxonomique (de quels organismes ces transcrits proviennent-ils ?). C’est le « qui fait quoi », détaillé dans (Carradec et al., 2018).
En parallèle, le séquençage métagénomique rend compte de l’ensemble des gènes non pas exprimés, mais présents. Ce type de séquençage informe sur le potentiel génétique de la communauté, c’est le « qui peut faire quoi », mais ne le fait pas forcément. Cette approche au potentiel colossal se confronte à de nombreuses limitations techniques. À l’époque de Tara Océans, la taille des fragments d’ADN pouvant être séquencés à haut débit dépassait à peine les 500 paires de base. Si l’on considère par exemple le génome d’une diatomée (phytoplancton abondant), qui fait environ 30 millions de paires de bases, cela signifie qu’il faut séquencer au minimum 60 000 fragments d’ADN pour reconstituer son génome complet. En pratique, les analyses ne sont jamais réalisées sur un seul individu, notamment parce qu’il faut disposer de fragments d’ADN qui se chevauchent pour pouvoir reconstituer l’ordre des séquences. En réalité, le séquençage est donc réalisé à partir d’un mélange de fragments d’ADN provenant d’individus différents. Il n’est donc pas possible de se limiter au séquençage de 60 000 fragments au hasard, car sinon certaines parties du génome de notre diatomée auront été séquencées plusieurs fois, mais d’autres aucunes et le génome reconstitué comportera donc des « trous ». Pour éviter ce problème, on séquence l’équivalent non pas d’un seul génome (60 000 fragments) mais, par exemple, de vingt génomes (donc 1 200 000 fragments !). À l’époque de Tara Océans, séquencer les génomes de plusieurs centaines ou milliers d’espèces présentes dans un échantillon d’eau semblait une tâche démesurée. Les défis en termes d’assemblage et d’annotation sont colossaux, mais des progrès sont réalisés chaque année.
Grands principes d’échantillonnage
Pour chacune des 210 stations de l’expédition Tara Océans, l’échantillonnage s’est fait à deux profondeurs. La première est la couche d’eau de surface (SUR), définie comme la couche de trois à sept mètres sous la surface. La seconde est la couche de « deep chlorophyll maximum » (DCM) qui correspond à la zone d’abondance maximale du plancton photosynthétique, déterminée grâce à la mesure de la concentration en chlorophylle par fluorimétrie.
Pour chaque station, l’échantillonnage s’est fait sur plusieurs fractions de taille. Le plancton prélevé pendant Tara Océans recouvre six ordres de grandeur en termes de taille qui correspondent aux virus, virus géants (girus), procaryotes (bactéries et archées), eucaryotes unicellulaires (protistes), et eucaryotes pluricellulaires (comme les copépodes). Les cellules des protistes mesurent entre 0,8 et 2000 microns. Des filets ont été utilisés afin de créer plusieurs fractions de taille : 0,8-5 microns, 5-20 microns, 20-180 microns, 180-2000 microns.
Pour chaque station, une analyse morphologique a été réalisée pour différentes classes d’organismes. D’une part, des systèmes de reconnaissance automatisés (le FlowCam et le ZooScan) permettent des mesures quantitatives de la biodiversité des organismes allant de 20 microns à quelques centimètres. D’autre part, la microscopie confocale 3D et la microscopie électronique à transmission permettent des analyses ultrastructurales détaillées des petits protistes.
Le projet Tara Océans a utilisé de nombreuses nouvelles technologies et outils d’analyses pour établir le premier effort de collecte de données à l’échelle planétaire qui couple biogéographie, écologie, génétique et morphologie, regroupant une communauté internationale de scientifiques issus de disciplines bien différentes : écologistes marins, microbiologistes, océanographes, statisticiens, biogéochimistes, informaticiens, ingénieurs. Cinq articles fondateurs ont été publiés dans le journal Science en mai 2015, et plus de 75 papiers scientifiques ont été publiés depuis le début de l’aventure.
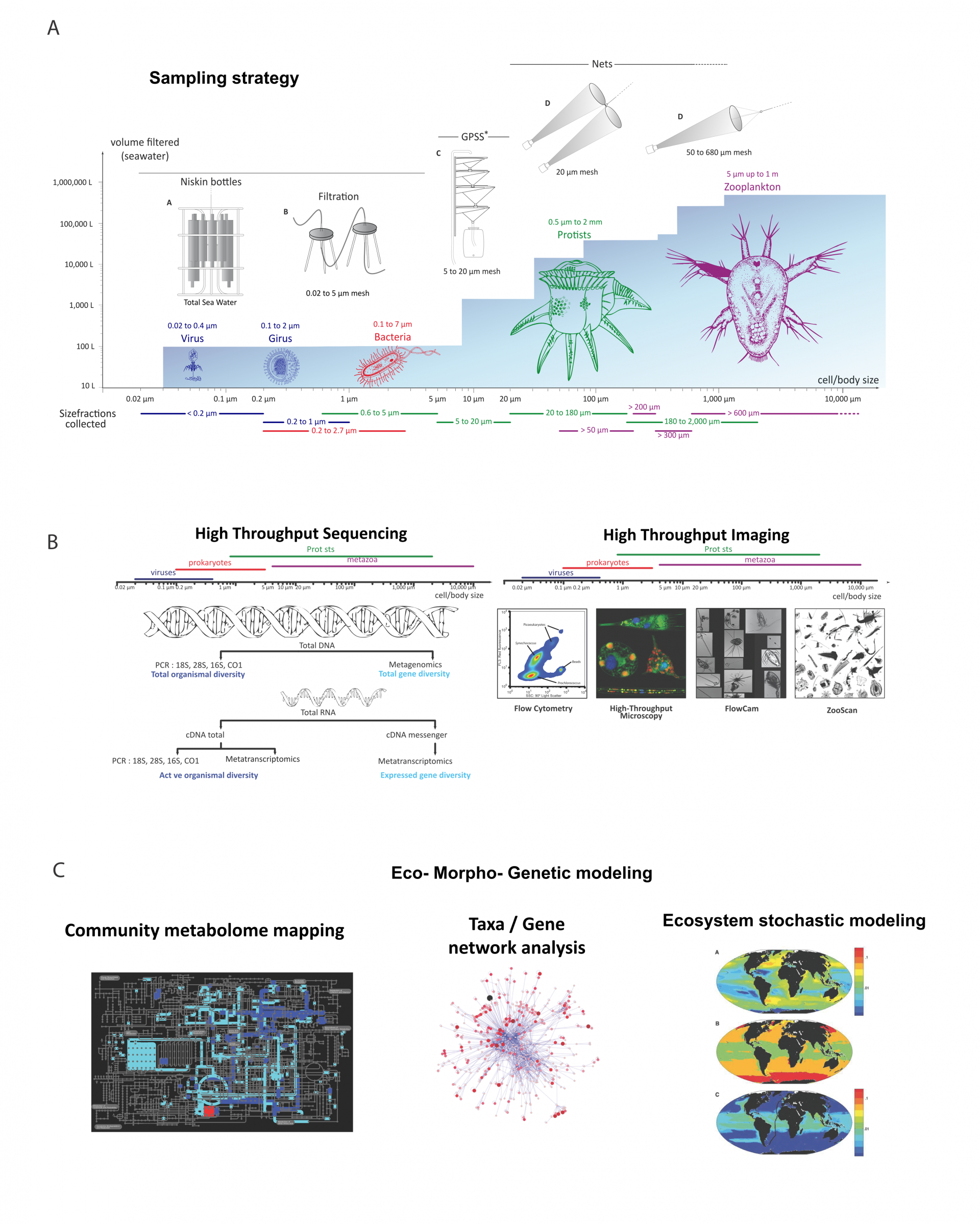
(A) Méthode pour échantillonner des organismes par fractions de taille. Le fond bleu représente le volume d’eau filtré afin d’obtenir suffisamment de biomasse pour l’analyse. (B) Méthode de traitement des échantillons : les images à droite proviennent de Tara Océans. (C) Séquençage à haut débit et imagerie quantitative apportent des données sur l’évolution, le métabolisme et les interactions entre les organismes, et permettent de reconstruire les métabolomes de la communauté, des réseaux de gène et d’organismes, ainsi que des modèles de distribution spatiale d’espèces.
Les premiers résultats en génétique de Tara Océans
Les éléments de cette partie sont issus d’un communiqué de presse de Tara Océans. Chaque sous-partie correspond au compte-rendu d’un article de recherche, dont la référence est donnée.
La découverte de plus de 100 000 types de protistes dans le plancton mondial enrichit notre compréhension des écosystèmes marins
Après trois ans de navigation et d’étude des zones baignées de lumière des océans planétaires, les chercheurs ont dévoilé une diversité insoupçonnée chez les organismes unicellulaires eucaryotes aussi appelés protistes. Le séquençage de près d’un milliard de codes-barres génétiques a montré que les protistes sont largement plus diversifiés que les bactéries ou les animaux, et que la plupart d’entre eux appartiennent à des groupes peu connus de parasites, de symbiontes, et de prédateurs en tout genre. Ces résultats changent radicalement notre vision de la diversité biologique et fonctionnelle du plancton mondial, écosystème-clef pour le fonctionnement de notre biosphère.
Dans cette étude, les chercheurs ont déchiffré et analysé près d’un milliard de séquences d’ADN ribosomique, issues de 334 sites d’échantillonnage, qui correspondent à des marqueurs de la biodiversité des eucaryotes, des plus petits organismes unicellulaires (<1 micron) aux animaux planctoniques de quelques millimètres.
La grande quantité de code-barres génétiques générés a tout d’abord permis de caractériser la quasi-totalité des espèces eucaryotes du plancton de la zone photique analysée. 150 000 types génétiques de plancton eucaryote ont été mis en évidence, ce qui représente une diversité insoupçonnée par rapport aux 11 000 espèces décrites jusqu’à présent. La grande majorité des types génétiques répertoriés n’a pas de référent proche dans les bases de données génétiques actuelles, démontrant que ces organismes sont pour la plupart non-répertoriés et incultivables. Un tiers de la diversité génétique n’a même pu être associé à aucune des grandes lignées eucaryotes reconnues aujourd’hui.
Parmi les types génétiques pouvant être classés dans l’arbre de la vie eucaryote, la plupart correspondent à des organismes unicellulaires (protistes), avec une diversité phénoménale de parasites, d’espèces symbiotiques, et de prédateurs en tout genre. Les organismes photosynthétiques sont, quant à eux, bien moins diversifiés, plus petits, et représenteraient une biomasse largement plus faible.
Référence : Eukaryotic plankton diversity in the sunlit ocean, C. de Vargas, S. Audic, N. Henry et al., Science, 22 mai 2015
Structure et fonction du microbiome océanique
Les virus et les microorganismes ([de taille inférieure à] 3 micromètres) dominent numériquement les écosystèmes planctoniques marins avec 10 à 100 milliards de cellules dans chaque millilitre d’eau de mer. Leur rôle majeur dans les grands processus biogéochimiques est bien connu et il est donc important de cataloguer leur diversité biologique et fonctionnelle et comprendre comment ils sont affectés par leur environnement. Ces questions ont été abordées pour la première fois à l’échelle planétaire grâce à la métagénomique, c’est-à-dire au séquençage massif du matériel génétique issu de communautés entières de microorganismes. Les communautés de microorganismes de tailles variées et vivant à différentes profondeurs ont été prélevés dans l’ensemble des océans planétaires et des relevés de plusieurs paramètres physicochimiques de leur environnement ont été effectués en parallèle.
La quantité d’ADN séquencé correspond à environ deux millions de génomes bactériens, ou à peu près deux mille génomes humains. Le catalogue de gènes issu de ces travaux comprend 40 millions de gènes de virus, de procaryotes et de pico-eucaryotes marins, nouveaux pour la plupart (> 80 %). Il permet de cartographier la diversité fonctionnelle de ces microorganismes océaniques et constitue une ressource fondamentale pour de nombreuses études scientifiques à venir.
Une première analyse a par exemple permis de dégager quels sont les paramètres environnementaux qui influencent la formation des communautés microbiennes dans la zone baignée de lumière des océans et d’identifier la température comme un des facteurs les plus importants. Ces résultats impliquent que le réchauffement climatique pourrait avoir un fort impact sur ces communautés microbiennes, invisibles à l’œil nu, dont l’activité photosynthétique est à la base des chaînes alimentaires marines.
Par ailleurs, une comparaison entre les familles de gènes qui sont au cœur du fonctionnement des communautés microbiennes océaniques et celles présentes dans le système digestif humain a montré que plus de la moitié sont partagées, indiquant des principes communs de la vie microbienne dans ces deux écosystèmes très distincts.
Ce nouveau catalogue de gènes microbiens marins planétaires représente un outil indispensable pour mieux comprendre la biodiversité des microbes du plancton et de leurs fonctions, et ce plus particulièrement dans le contexte du changement climatique.
Référence : Structure and function of the global ocean microbiome, S. Sunagawa, L.P. Coelho, S. Chaffron, et al., Science, 22 mai 2015.
Cartographie des interactions planctoniques : le rôle majeur des parasites
Si la biodiversité des communautés microscopiques qui sont à la base de la vie marine commence à être bien répertoriée, la structure et la dynamique de ces communautés sont encore mal connues. Les microorganismes planctoniques interagissent de différentes façons (compétition, collaboration, prédation, symbiose, parasitisme) et forment de gigantesques chaînes alimentaires qui affectent des processus majeurs tels que la séquestration du carbone et la photosynthèse. Cependant la plupart de ces interactions étaient jusqu’à présent largement inconnues.
Dans cette étude, les chercheurs se sont intéressés à la cartographie des réseaux d’interactions entre espèces de plancton dans la zone photique, et à la façon dont la structure et la composition des communautés planctoniques sont façonnées par des facteurs biotiques (interactions entre espèces) et abiotiques (conditions environnementales et disponibilité en nutriments). Ils ont développé de nouveaux modèles permettant de prédire les interactions au sein des communautés planctoniques. Grâce à l’analyse des échantillons prélevés lors de l’expédition avec des techniques de microscopie de pointe, ils ont ensuite confirmé que les interactions prédites par les modèles étaient bien réelles dans les communautés observées.
Les analyses dites de « réseaux de co-abondances » montrent que les associations au sein du plancton ne sont pas distribuées de façon aléatoire et que les facteurs abiotiques ont des effets plus limités que prévu sur la structure des communautés. Ils soulignent le rôle des interactions biotiques (relation de type top-down, où les ressources sont régulées par les consommateurs) dans la zone supérieure de l’océan et notamment du parasitisme. Les interactions parasitiques sont en effet les plus abondantes dans le réseau généré par ordinateur et ont également été observées de façon répétée dans les échantillons. La fréquence élevée du parasitisme chez les microorganismes océaniques constitue l’observation la plus importante de cette étude et semble indiquer que les parasites jouent un rôle majeur et largement sous-estimé dans l’écologie du plancton marin.
Cette première cartographie globale des interactions planctoniques constitue la matière première qui permettra de savoir comment les symbiontes, pathogènes, prédateurs et parasites interagissent avec leurs organismes cibles, d’élucider le fonctionnement des chaînes alimentaires et la circulation des flux de nutriments et d’énergie, pour à terme mieux comprendre et prédire la dynamique des écosystèmes océaniques.
Référence : Determinants of community structure in the global plankton interactome , G. Lima-Mendez, K. Faust, N. Henry et al., Science, 22 mai 2015.
Conclusion
À ce jour (7 décembre 2018), 68 publications émanent directement de l’analyse des données de Tara Océans. Qu’elles s’intéressent à la diversité des virus, des bactéries, du phytoplancton, au lien entre cet écosystème planctonique et l’export de carbone ou encore à l’évolution de ces micro-organismes dans nos océans, l’avalanche de résultats démontre que ces données sont une mine d’or. Les études représentent aussi bien une démarche scientifique traditionnelle, avec formulation d’hypothèses et validation expérimentale, que de nouvelles approches dites « data driven ». L’idée de celles-ci est de laisser les données parler par elles-mêmes pour nous indiquer des pistes de recherches auxquelles nous n’aurions pas forcément pensé, tant la nature est plus créative que le cerveau humain.
S’engager dans une telle aventure, avec une manne de données si colossale et complexe, suppose de repenser les façons de faire de la recherche scientifique au XXIᵉ siècle. D’une part, au sein du consortium, cela impose d’être collaboratif et interdisciplinaire. La force de ce jeu de données réside dans sa capacité à coupler génétique, morphologie, biogéochimie et physique de l’océan à grande échelle spatiale. Il impose aux scientifiques d’oublier un instant leur jargon respectif et d’établir un langage commun pour exprimer simplement leurs idées et aboutir à des questionnements qui gomment les frontières disciplinaires. D’autre part, il serait illusoire de penser un instant qu’un groupe de 100 scientifiques suffirait à exploiter ces données dans leur intégralité. Ainsi, ces ressources sont rendues accessibles, facilement utilisables et compréhensibles, afin que chaque scientifique où qu’il soit puisse les analyser librement. Dans une époque où la donnée est plus que jamais monétisée et la science de plus en plus appliquée, Tara Océans nage à contre-courant en donnant librement accès, au nom de l’amélioration de nos connaissances, à la diversité du plus grand écosystème de la planète : l’océan.
Références
- Blaxter, M., Mann, J., Chapman, T., Thomas, F., Whitton, C., Floyd, R., and Eyualem, A. (2005) Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. 360: 1935–1943.
- Carradec, Q., Pelletier, E., Da Silva, C., Alberti, A., Seeleuthner, Y., Blanc-Mathieu, R., et al. (2018) A global ocean atlas of eukaryotic genes. Nat. Commun. 9: 1038.
- Field, C.B. (1998) Primary Production of the Biosphere: Integrating Terrestrial and Oceanic Components. Science (80-. ). 281: 237–240.
- Karsenti, E., Acinas, S.G., Bork, P., Bowler, C., De Vargas, C., Raes, J., et al. (2011) A holistic approach to marine eco-systems biology. PLoS Biol. 9: e1001177.
- Kress, W.J., Erickson, D.L., Uriarte, M., and Garcı, C. (2015) DNA barcodes for ecology , evolution , and conservation. 30.
- Logares, R., Sunagawa, S., Salazar, G., Cornejo‐Castillo, F.M., Ferrera, I., Sarmento, H., et al. (2014) Using mitags to explore microbial communities. Env. Microbiol 2659–2671.
- Taberlet, P., Coissac, E., Pompagnon, F., Brochmann, C., and Willerslev, E. (2012) Towards next‐generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21: 2045–2050.
- Valentini, A., Pompanon, F., and Taberlet, P. (2009) DNA Barcoding for Ecologists. Trends Ecol. Evol. 24: 110–17.
- Zagoskin, M., Lazareva, V., Grishanin, A., and Mukha, D. (2014) Phylogenetic information content of Copepoda ribosomal DNA repeat units: ITS1 and ITS2 impact. Biomed Res Int.



