
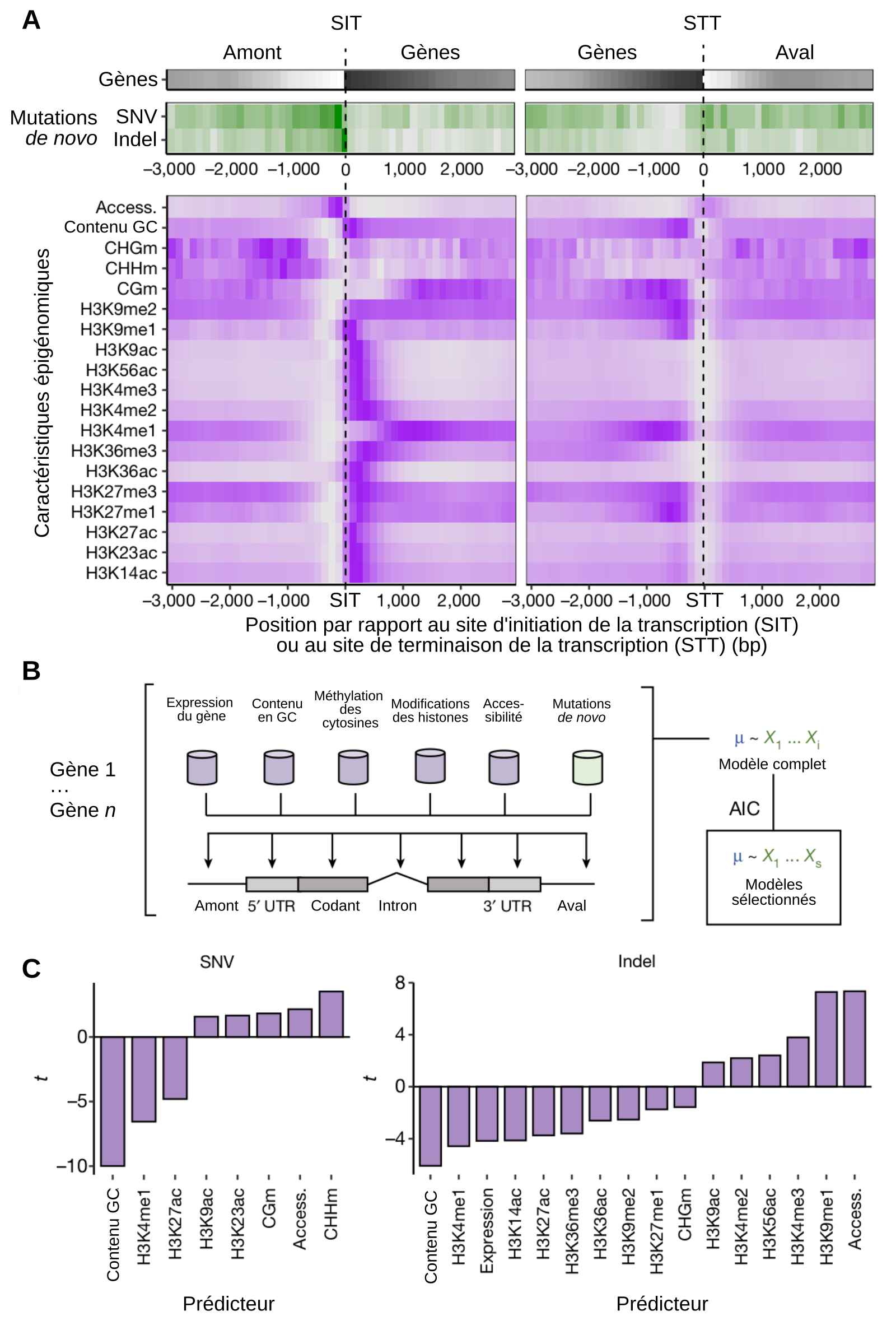
A. Densité des séquences transcrites (en noir1), des mutations observées (en vert), et de marques épigénétiques (en violet) dans tout le génome, positionnés relativement aux sites d’initiation (SIT) et de terminaison (STT) de la transcription. Deux types de mutations sont identifiés : les SNV (single nucleotide variation), qui correspondent aux substitutions d’un nucléotide par un autre, et les indels, pour insertion/déletion. Les notations CHGm, CHHm et CGm correspondent à des méthylations de cytosines dans différents contextes : C = cytosine, H = adénine, cytosine ou thymine, G = guanine. Les autres caractéristiques épigénomiques indiquées correspondent à des modifications d’histones. Il s’agit de mono, di ou triméthylations (me1, 2, 3) ou d’acétylations (ac) sur différentes lysines (K) de l’histone H3. Par exemple, H3K36me3 signifie que la lysine 36 de l’histone H3 est triplement méthylée.
B. Modèle prédictif des probabilités de mutations, établi à partir des données exposées en A. Le modèle est sélectionné parmi plusieurs modèles linéaires par comparaison des AIC (Akaike Information Criterion).
C. Valeurs statistiques liées au modèle prédictif. Une valeur positive de t indique une corrélation positive entre le type de mutation observé et une caractéristique épigénétique donnée. Par exemple, les méthylations des cytosines sur des séquences de type CHH (CHHm) augmentent la probabilité des mutations de type SNV, tandis que les méthylations de la lysine 4 de l’histone H3 (H3K4me1) diminuent la probabilité des mutations de type insertion-délétion.
1La densité des séquences transcrites est représentée avec un dégradé allant du noir au blanc. Si les régions en amont du site d'initiation de la transcription, et celles en aval du site de terminaison, ne sont pas entièrement blanches, c'est parce que ces régions contiennent également des séquences transcrites. Autrement dit, toutes les séquences transcrites d'A. thaliana ne sont pas forcément séparées par au moins 3000 pb.


{kind=link}