

Cet article présente le principe et les différentes techniques permettant de réaliser une électrophorèse, ainsi que leurs applications (ADN, protéines...) et limites.
Principe de la technique
L’électrophorèse est une technique séparative. Elle est utilisée le plus souvent dans un but analytique mais également parfois pour purifier des molécules solubles. Elle n’est donc pas adaptée à la séparation des lipides. Le principe consiste à soumettre un mélange de molécules à un champ électrique ce qui entraîne la migration des molécules chargées. En fonction de différents paramètres (charge, masse, forme, nature du support, conditions physico-chimiques) la vitesse de migration va être variable, ce qui permet la séparation des différentes molécules. A partir de ce principe général, il existe plusieurs variantes de cette technique adaptées à différentes situations. Sans prétendre à l’exhaustivité, cet article présente les principales sortes d’électrophorèse, leurs intérêt et limite.
Le support
La migration des molécules doit au minimum être guidée. Elle peut ainsi être réalisée dans un fin tuyau, on parle alors d’électrophorèse en veine liquide. Cette technique est assez complexe à mettre en œuvre et ne permet pas une séparation optimale des différents constituants c’est pourquoi elle n’est pas beaucoup utilisée. Notons qu’il ne faut pas confondre l’électrophorèse en veine liquide avec la chromatographie liquide à haute performance(HPLC) qui utilise également des capillaires. En effet, dans les deux cas le facteur utilisé pour la séparation les molécules est totalement différent (pour plus d'information sur l'HPLC, voir l'article sur La chromatographie).
Mais dans la grande majorité des cas, l’électrophorèse utilise un support. Les plus courants sont le papier, l’acétate de cellulose, les gels d’agarose et les gels de polyacrylamide. Idéalement, le support d’une électrophorèse devrait être parfaitement inerte chimiquement, contrairement à ce qui se passe pour certains types de chromatographie. Seul un effet mécanique ralentissant la progression des molécules est recherché, ce qui permet d’améliorer la séparation. Bien entendu, cet idéal n’est jamais totalement atteint, et c’est particulièrement vrai pour le papier et l’acétate de cellulose. De ce fait, l’électrophorèse sur ces supports met à profit les interactions différentielles qui existent entre certaines classes de molécules et le support pour leur séparation (exemple de la séparation des protéines sériques), mais cela ne permet pas une très bonne résolution. Par ailleurs, ces supports se distinguent des gels d’agarose et de polyacrylamide en ce sens que la migration s’effectue en surface de la bande de papier ou d’acétate de cellulose (qui présente une pellicule de solution tampon), alors qu’elle s’effectue dans l’épaisseur des gels.
Le choix du support est dicté par la nature des molécules à séparer. Ainsi, les gels forment un maillage tridimensionnel. Les molécules doivent donc progresser dans les pores ; les espaces libres situés entre les mailles du filet. On conçoit aisément que plus les molécules seront encombrantes, plus leur progression sera difficile donc lente. Il arrive même un moment où leur encombrement ne leur permet plus de progresser du tout. Au-delà de cette limite, il n’y aura donc aucune séparation. A l’inverse, des molécules très petites, comparées aux pores, ne seront pratiquement pas freinées, et par suite mal séparées. En fonction de la taille des pores, on peut donc efficacement séparer des molécules situées dans une fourchette approximative de masses moléculaires. On peut, bien sûr, adapter la réticulation du gel à l’échantillon à traiter en choisissant la concentration du produit (voir Fig. 1). Cela suppose d’avoir une idée de la nature des molécules que l’on souhaite séparer. Dans le cas contraire, on peut faire plusieurs gels différents, voire faire un gel présentant un gradient de concentration (en polyacrylamide).
| Pourcentage d'acrylamide | Gamme de séparation en kDa |
| 7,5% 10% 12% 15% |
45-400 22-300 13-200 2,5-100 |
| La réalisation d'un gel de polyacrylamide se fait par polymérisation d'une solution d'acrylamide (qui forme des chaines) par du bisacrylamide qui lie ces chaînes entre elles (on parle de pontage). En fonction de la concentration de départ de la solution d'acrylamide, le réseau obtenu sera plus ou moins réticulé, permettant la séparation de molécules plus ou moins massives. | |
D’une manière générale, l’agarose forme des gels dont la réticulation est assez faible, permettant la séparation de molécules de très hautes masses moléculaires. Ils sont principalement utilisés pour séparer des molécules d’ADN ou d’ARN.
L’acrylamide-bisacrylamide forme des réseaux beaucoup plus réticulés, ils sont donc indiqués pour séparer des molécules de masses moléculaires moins élevées, typiquement les protéines. Notons que des fragments d’ADN de tailles limitées peuvent nécessiter l’utilisation de gels d’acrylamide plutôt que d’agarose. C’est en particulier le cas des fragments utilisés pour le séquençage qui ne dépassent généralement pas quelques centaines de nucléotides et dont le gel doit permettre de séparer des fragments dont la taille diffère d’un seul nucléotide (voir animation Fig. 2). Pour les molécules les plus petites, comme les acides aminés, les gels ne conviennent pas pour leur séparation.
L’électrophorèse sur papier ou acétate de cellulose ne présente évidemment pas les mêmes contraintes puisque les molécules restent en surface. Il est en particulier possible de séparer les molécules les plus petites. En revanche, la résolution est assez limitée et il peut être préférable d’utiliser d’autres techniques de séparation telle que la chromatographie.
L’électrophorèse en conditions non dénaturantes
Dans l’électrophorèse en conditions non dénaturantes, les molécules sont séparées dans leur état le plus proche possible de leur état natif. Les étapes préalables permettant l’obtention de la solution contenant les molécules à séparer doivent éviter les conditions dénaturantes. Sauf cas particulier, il faut donc éviter les pH élevés ou au contraire très bas, les traitements à des températures élevées, les hautes forces ioniques (fortes concentrations en sel) et les agents chimiques dénaturants qui seront évoqués au paragraphe suivant. Les résultats obtenus sont toujours délicats à interpréter concernant la structure des molécules, car la séparation se fait en fonction de nombreux critères. Pour un support et un voltage donnés, la vitesse de migration dépendra de :
-
la charge native de la molécule. Plus la charge est importante, plus la vitesse de migration sera importante. Signalons que cette charge native peut varier en fonction du pH, qui est le plus souvent fixé par l’expérimentateur au moyen de l’utilisation d’une solution tampon. Signalons également que selon le signe de la charge native, les molécules pourront migrer vers l’anode (molécules chargées négativement) ou vers la cathode (molécules chargées positivement) et que, pour les protéines, il convient donc de faire le dépôt à mi-chemin des deux électrodes, sauf à vouloir séparer une protéine dont on connaît la charge. Enfin, toutes les molécules non chargées ne seront pas séparées puisqu’elles ne migreront pas du tout. Cette question ne se pose pas pour les acides nucléiques (ADN et ARN) ; les acides phosphoriques qu’ils contiennent leur donnant une charge toujours négative.
-
la masse moléculaire. Plus la masse moléculaire est importante, plus la vitesse de migration de la molécule sera faible. Rappelons que le choix du support est essentiel. En effet, un support mal choisi peut entraîner une absence de migration (molécule trop grosse pour le maillage du support) ou au contraire une mauvaise, voire aucune séparation (molécule trop petite pour le maillage du support).
-
la structure tridimensionnelle. En effet, à masse moléculaire égale, une molécule pourra, selon sa forme, être plus ou moins ralentie dans sa progression par le support. Par exemple, une molécule de structure globulaire sera moins ralentie qu’une molécule de structure fibreuse. On citera comme meilleur exemple la séparation de molécules d’ADN circulaires identiques (donc de même masse moléculaire) mais présentant des états de surenroulement différents (ce qui influence l’état de compaction de l’ADN).
Cette technique présente certains avantages comme celui de permettre la séparation de molécules multimériques entières. En revanche, outre l’interprétation délicate, il est fréquent que les molécules présentes dans l’échantillon soumis à électrophorèse s’agrègent entre elles, formant de très gros ensembles impossibles à séparer.
Pour terminer, citons une variante de cette technique qui consiste à utiliser un gel dont le pH varie selon un gradient entre les deux électrodes. Chaque molécule chargée verra sa charge varier au fur et à mesure de son déplacement (le pH étant différent en chaque point du gel) jusqu’au moment où la molécule va atteindre son point isoélectrique (voir article sur Les acides aminés). N’étant plus chargée, elle ne va plus se déplacer : on est à l’équilibre. On sépare donc les différentes molécules en fonction de leur point isoélectrique d’où le nom d’isoélectrofocalisation.
L’électrophorèse en conditions dénaturantes
Comme son nom l’indique, dans cette variante, les molécules sont soumises à un traitement dénaturant préalablement à leur séparation électrophorétique, détruisant la structure tridimensionnelle native. Il existe différentes méthodes pour dénaturer les molécules. Les plus classiques sont les traitements thermiques, les hautes forces ioniques qui vont perturber les liaisons faibles qui participent au repliement des molécules (liaisons hydrogènes, liaisons électrostatiques), et, bien sûr, l’utilisation d’agents dénaturants comme l’urée ou le SDS (sodium dodécyl sulfate). Ce dernier est un détergent très utilisé pour l’électrophorèse de protéines car il possède des caractéristiques particulièrement intéressantes. Non seulement, il dénature les protéines, mais il se fixe dessus avec une densité linéaire approximativement constante, c’est-à-dire que le nombre de molécules de SDS qui se fixe sur une protéine est approximativement proportionnel au nombre d’acides aminés qui la composent, donc à sa masse moléculaire. Or, le SDS est une molécule chargée négativement. En sa présence, toutes les protéines vont donc adopter la même forme (déroulées) avec une charge négative proportionnelle à la masse moléculaire. On estime ainsi qu’il se fixe en moyenne environ deux molécules de SDS par acide aminé. Sauf cas particulier, la charge native est faible donc négligeable par rapport aux charges portées par le SDS. Il en résulte qu’en présence de SDS la seule variable qui différencie les protéines est la masse moléculaire. La forme n’intervient plus (puisque les protéines sont dénaturées) et la charge native non plus (puisque les charges apportées par le SDS sont largement plus nombreuses). La séparation se fait donc uniquement en fonction de la masse moléculaire. Par voie de conséquence, l’interprétation des résultats obtenus est généralement beaucoup plus simple que pour une électrophorèse en condition non dénaturante. Autre avantage, l’utilisation d’un détergent permet de solubiliser les protéines hydrophobes, et en particulier les protéines membranaires, qui ne peuvent pas être séparées par électrophorèse en conditions non dénaturantes. Comme le support le plus adapté pour séparer les protéines est le gel de polyacrylamide (compte tenu du fait qu’une protéine typique fait autour de 50 kDa) on parle couramment de SDS-PAGE (pour PolyAcrylamide Gel Electrophoresis ou gel de polyacrylamide en présence de SDS). L’urée est, elle, généralement utilisée pour dénaturer l’ADN.
Un autre agent chimique couramment utilisé est le β-mercaptoéthanol. C’est un agent réducteur qui a pour propriété de réduire les ponts disulfures des protéines, donc de détruire ces pontages covalents qui ne sont pas cassés par le SDS. Cela permet de séparer les différents polypeptides reliés par de tels ponts au sein d’une même protéine. La comparaison de SDS-PAGEs avec et sans β-mercaptoéthanol d’un même échantillon est donc riche d’informations sur la structure quaternaire des protéines (voir Fig. 3).

Les mêmes échantillons ont été soumis à séparation électrophorétique sur gel de polyacrylamide (7,5 %)-bisacrylamide (0,2 %), après dénaturation par du SDS, en présence (A, B, C) ou en absence (D, E) de β-mercaptoéthanol. Le gel est coloré au bleu de Coomassie. On constate que les masses moléculaires apparentes observées sont plus élevées en absence (170 ou 180 kDa) qu’en présence de β-mercaptoéthanol (bande diffuse à 75-80 ou 80kDa). On peut en déduire que la molécule isolée est composée de deux polypeptides reliés par un ou plusieurs pont(s) disulfure(s) interchaînes. Signalons que la protéine native est composée de deux unités de 170/180 kDa qui sont dissociées par le SDS. Une électrophorèse en conditions non dénaturantes aurait donc donné une bande au-delà des 300 kDa
Colonne A : marqueurs de masse moléculaire ; colonnes B et E : DBH (dopamine-β-hydroxylase) purifiées à partir de phéochromocytomes de rat ; C et D : DBH purifiées à partir de phéochromocytomes humains.
L’électrophorèse bidimensionnelle
L’électrophorèse bidimensionnelle ne fait que reprendre les deux types d’électrophorèses présentées dans les deux paragraphes précédents, mais appliquées successivement sur le même gel selon deux orientations différentes.
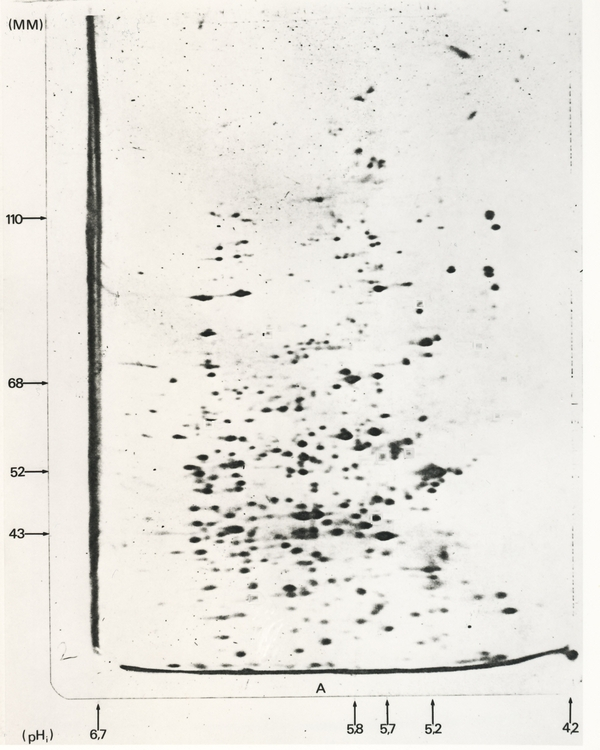
La migration initiale a été réalisée en conditions dénaturantes (SDS), en présence de β-mercaptoéthanol. La révélation est obtenue par autoradiographie, les protéines produites par l’ovocyte ayant été marquées au moment de leur synthèse par l’utilisation de méthionine35S (marquage métabolique). L’axe des abscisses donne le point isoélectrique des différentes protéines séparées, l’axe des ordonnées leur masse moléculaire apparente. On identifie de très nombreux spots dont certains ont une même masse moléculaire apparente (situés sur une même ligne horizontale). Un gel de ce type permet donc de séparer de très nombreuses protéines en une seule étape.
La première étape consiste à réaliser une isoélectrofocalisation, c’est-à-dire à séparer les molécules selon leur point isoélectrique. Afin d’éviter que les différents polypeptides qui composent une même molécule soient tous regroupés au niveau du même point isoélectrique, correspondant au point isoélectrique global de la protéine, les échantillons sont d’abord traités avec du β-mercaptoéthanol (qui réduit et donc « casse » les ponts disulfures) ainsi qu’avec de l’urée, un agent dénaturant. Ce traitement laisse néanmoins inchangées les charges intrinsèques des différents polypeptides. Dans un échantillon complexe, comme un broyat total d’un tissu, le nombre de protéines est tel (plusieurs milliers), que de nombreuses molécules possèdent un point isoélectrique identique, et ne sont donc pas séparées les unes des autres lors de cette première étape.
Une deuxième migration, réalisée dans une direction perpendiculaire à la première (d’où le nom d’électrophorèse bidimensionnelle), permet ensuite la séparation des protéines en fonction de leur masse moléculaire. Cette seconde migration correspond à un SDS-PAGE à l’issue duquel de nombreux points peuvent être individualisés sur le gel (Figure 2). L’électrophorèse bidimensionnelle présente ainsi un pouvoir de résolution plus important qu’une simple électrophorèse unidimensionnelle.
Quelques techniques de révélation
Après séparation, les molécules doivent pouvoir être localisées. Il est évident que la méthode de révélation dépendra de la nature des molécules à mettre en évidence. Mais un souci est à prendre en compte : il faut rapidement stabiliser l’emplacement des molécules. En effet, dès la fin de l’électrophorèse, la diffusion reprend ses droits, avec pour conséquence une perte de résolution (les molécules regroupées à un endroit précis ayant tendance à se disperser). Pour cela on peut fixer les molécules sur leur support en plongeant le gel dans une solution contenant un mélange de méthanol et d’acide acétique, ce qui a pour effet de dénaturer définitivement les protéines et de les fixer au support. Ultérieurement, on peut aussi déshydrater le gel pour une conservation de longue durée. Mais on peut également transférer les molécules sur un autre support sur lequel elles se fixeront fortement. C’est ce que l’on appelle un blot. Ce second support est une membrane de nitrocellulose, un papier établissant cette fois des interactions chimiques avec les molécules. Une fois les molécules transférées, on peut leur faire subir différents traitements, ce qui n’entraînera pas leur décrochage.
Pour l’ADN, la méthode la plus classique est d’utiliser un agent intercalant fluorescent tel que le bromure d’éthidium. Cette molécule plate va s’intercaler entre les bases des nucléotides. Lorsqu’on éclaire cette molécule avec des UV à courtes longueurs d’ondes (autour de 300 nm), elle réémet de la lumière visible rouge orangée. On peut observer directement la lumière produite ou faire une photographie. Signalons qu’il faut utiliser des protections adaptées lors de cette révélation. En effet, une longueur d’onde de l’ordre de 300 nm correspond à des rayons UV très énergétiques. D’autre part, les lampes utilisées sont généralement d’assez forte puissance. La peau et surtout les yeux doivent donc être protégés : port de vêtements longs, de gants et d’un masque transparent anti-UV (des lunettes anti-UV protègent les yeux mais pas la peau du visage : bronzage « retour de ski » garanti en une minute). Les méthodes chimiques utilisant des colorants sont moins utilisées car ils sont moins sensibles.
Pour les protéines, il y a de multiples façons de faire. La plus simple est d’utiliser un colorant, le bleu de Coomassie, avant de déshydrater le gel. La technique est très simple (cinq minutes d’incubation dans une solution contenant le colorant) mais sa sensibilité reste modeste. On peut aussi utiliser une coloration beaucoup plus sensible au nitrate d’argent, mais cette méthode est plus longue et complexe à mettre en œuvre. Après transfert sur membrane de nitrocellulose on peut utiliser des méthodes indirectes basées sur la reconnaissance spécifique antigène-anticorps. On parle alors d’immunoblot. Une autre possibilité consiste à faire une autoradiographie, ceci dans le cas où des molécules comportant des atomes radioactifs sont présentes (marquages métaboliques par exemple). La figure 5 présente deux exemples de révélation d’un même échantillon.
Cette liste est loin d’épuiser la variété des techniques existantes, d’autres méthodes étant disponibles en fonction des particularités des molécules étudiées.
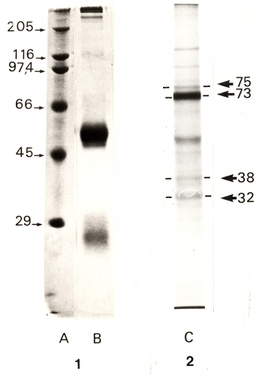
L’échantillon testé est le résultat d’une immunoprécipitation de DBH (dopamine-β-hydroxylase) humaine marquée lors de sa synthèse par l’utilisation de méthionine35S (ce marquage a été rendu possible car la protéine a été synthétisée dans des œufs de Xénope, suite à l’injection dans cet œuf de l’ARNm humain purifié codant pour la DBH). La séparation électrophorétique a été réalisée par SDS-PAGE en présence de β-mercaptoéthanol. L’image 1 montre le résultat de la coloration du gel au bleu de Coomassie et l’image 2 le résultat de l’autoradiographie du même gel. En coloration au bleu de Coomassie, on constate la présence d’une bande très importante de masse moléculaire apparente égale à 50 kDa environ. Cette bande correspond à la chaîne lourde des anticorps utilisés pour l’immunoprécipitation et ne correspond donc pas à la protéine d’intérêt (de même pour la bande d’environ 25 kDa de masse moléculaire apparente qui correspond à la chaîne légère des anticorps). En revanche, par autoradiographie, seules les molécules marquées sont révélées avec une bande largement majoritaire située à 73 kDa de masse moléculaire apparente, correspondant à la DBH (voir Fig. 3). On remarquera que cette bande est invisible sur le gel coloré au bleu de Coomassie, ce qui illustre bien la différence de sensibilité entre les méthodes de révélation disponibles. Les autres bandes observées (plus faibles) correspondent soit à des produits de dégradation, soit à d’autres protéines qui sont également reconnues par les anticorps utilisés, soit à des contaminants. En effet, étant donné la sensibilité de l’autoradiographie, il faut très peu de contaminants pour voir apparaître une bande.
Colonne A : marqueurs de masse moléculaire ; colonnes B et C : immunoprécipitation d’un surnageant de culture d’ovocytes injectés avec le l’ARNm humain de DBH, coloration au bleu de Coomassie (B) ou révélation par autoradiographie (C).


