
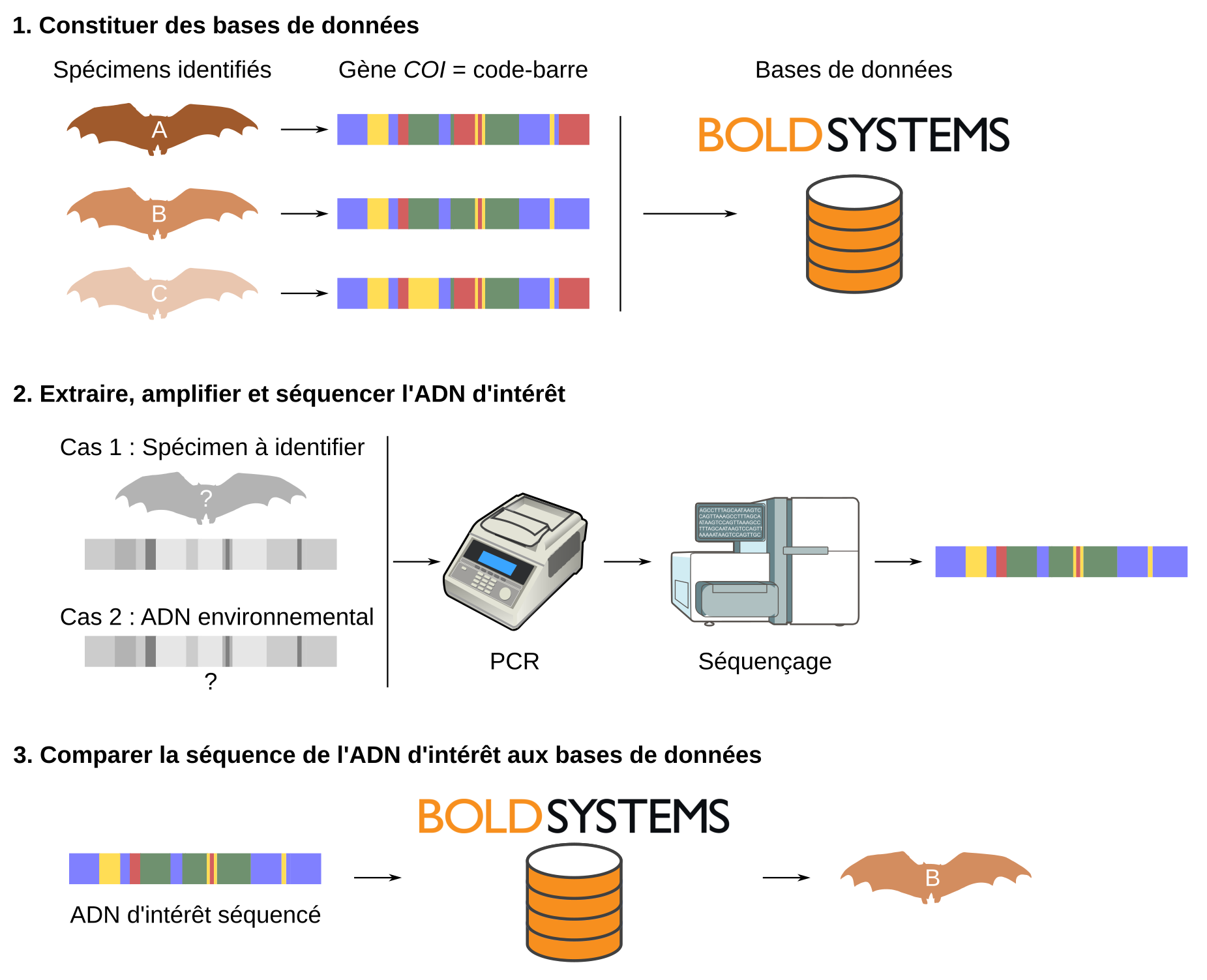
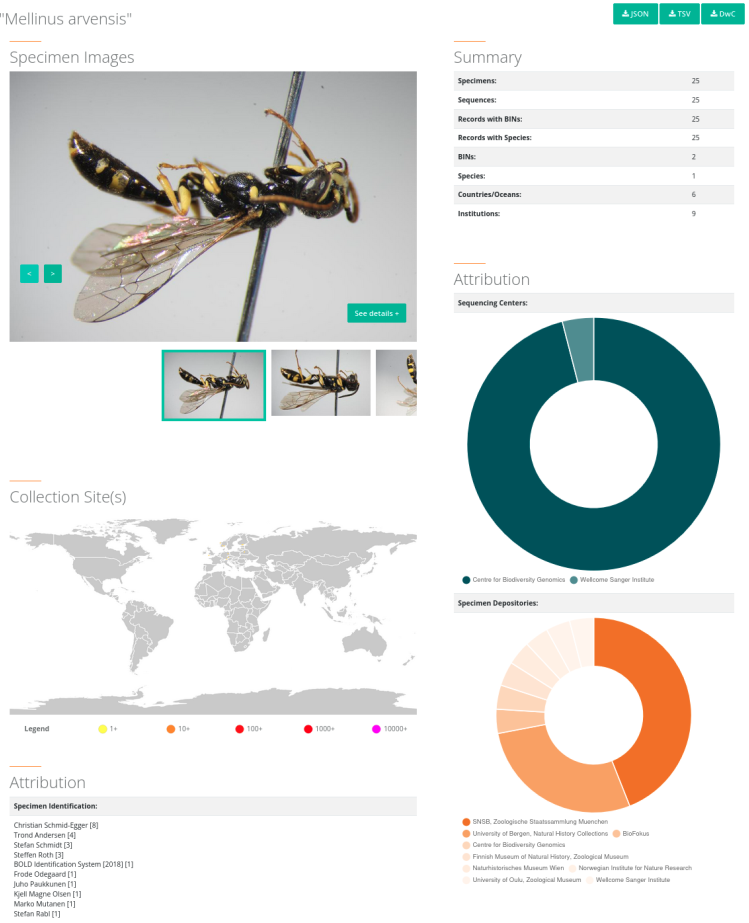
1. Pour pouvoir identifier des spécimens à partir de leur ADN, il est nécessaire de construire au préalable une base de données (comme la Barcode of Life Database, BOLD) dans laquelle des spécimens sont associés à des noms d’espèces et à un code-barre ADN. Chez les animaux, le code-barre utilisé est le gène mitochondrial COI, d’environ 600 paires de bases. En effet, ce gène présente une grande variabilité interspécifique, mais une faible variabilité intraspécifique. D’autres gènes peuvent être utilisés selon les taxons.
2. À l’étape suivante, le matériel de départ peut soit être un échantillon d’ADN issu d’un individu dont on souhaite connaître l’espèce, soit un échantillon d’ADN environnemental (eau, sol, fèces…) qu’on cherche à caractériser. Un échantillon d’ADN environnemental contient généralement des séquences issues de très nombreuses espèces, mais une seule est représentée ici, pour simplifier. Dans les deux cas de figure, l’ADN isolé est ensuite amplifié par PCR pour être présent en suffisamment grande quantité pour pouvoir être séquencé. L’amplification par PCR est réalisée grâce à des amorces spécifiques du gène utilisé comme code-barre. Le séquençage peut être réalisé avec la méthode de Sanger ou avec des méthodes de nouvelle génération (NGS) voire de troisième génération (TGS).
3. Une fois la séquence d’intérêt connue, il suffit de la comparer aux séquences de la base de données constituée à l’étape 1 pour identifier la séquence la plus ressemblante dans la base de données. Si celle-ci est suffisamment ressemblante (en général un seuil de similarité de 98 ou 99 % est retenu), cela permet d’identifier l’espèce à laquelle appartient l’ADN isolé à l’étape 2.
Source des images : Silhouette de chauve-souris :Ryan Kissinger pour NIAID NIH BioArt, domaine public ; Séquenceur de nouvelle génération : Ryan Kissinger pour NIAID NIH BioArt, domaine public ; Thermocycleur pour PCR : DBCLS, CC BY, Bioicons ; Base de données : gbrown, OpenClipart, CC0.


{kind=link}
{kind=link}
{kind=link}