Mis en évidence pour la première fois il y a trente ans seulement, le virus de l'hépatite C infecte 1 % de la population mondiale. Différentes molécules, interférant avec certaines étapes du cycle viral, ont été développées pour freiner le développement de la maladie. L'efficacité thérapeutique des traitements récents est telle qu'elle laisse espérer une prochaine éradication du virus.
L’hépatite C
Le virus de l’hépatite C (VHC) est un petit virus enveloppé à ARN simple brin. Du fait de sa variabilité génétique, on distingue différentes souches appelées génotypes (numérotés de 1 à 6), elles-mêmes subdivisées en sérotypes (notés a, b, c…). Il appartient à la famille des Flaviviridae et au genre Hepacivirus. Il est préférentiellement retrouvé dans les cellules hépatiques, mais peut être également détecté au sein de lymphocytes circulants [1]. Il est pathogène uniquement pour l’espèce humaine et ses proches cousins, les chimpanzés [2].
Après une contamination, 30 % des individus guérissent spontanément et 70 % des infections évoluent vers une forme chronique. 95 % de ces infections chroniques causent une inflammation du foie appelée hépatite, augmentant le risque de fibrose (remplacement des hépatocytes par du tissu cicatriciel) et pouvant aboutir à une cirrhose (stade très avancé de fibrose dans l’ensemble du foie) dans environ 20 % des cas. Chez certains patients, elles peuvent mener à des processus cancéreux, dont résultent des hépatocarcinomes [3] (liés à la cirrhose) et de rares lymphomes (dus à l’infection virale des lymphocytes B ou à la stimulation antigénique chronique).
Selon l’Organisation mondiale de la santé (OMS), l’hépatite C serait responsable d’au moins 30 % des décès par hépatite et toucherait environ 1 % de la population mondiale [4]. Le virus se transmet principalement par échange de sang, par exemple dans le cas de partage de seringues chez les toxicomanes ou de matériel souillé (rasoir, aiguille de tatouage…) ainsi que lors de soins médicaux et chirurgicaux non sécurisés (transfusions sanguines, facteurs anti-hémophiliques avant 1986…).
D’après l’étude des génomes des différentes souches du VHC, sa transmission interhumaine aurait commencé au début du XXe siècle, peut-être à partir de l’Asie du Sud-Est ou de l’Afrique de l’Ouest [5]. Sa propagation a été grandement accélérée au milieu du XXe siècle par les consommations de drogues administrées en intraveineuses et les soins hospitaliers nécessitants des transfusions [6]. L’épidémie est devenue importante entre les années 1940 et 1980, entraînant une grande diversification virale et l’apparition de différents génotypes dans les pays occidentaux [7]. Le virus a été identifié en 1989.
Découverte du virus
Dans les années 70, les hépatites secondaires aux transfusions sanguines étaient principalement dues à un petit virus enveloppé appelé « virus de l’hépatite non-A, non-B ». Cependant les techniques traditionnelles d’immunologie utilisées pour le virus de l’hépatite B (VHB) n’avaient pas réussi à mettre en évidence des antigènes ou des anticorps spécifiques du virus, peut-être à cause d’une trop grande dilution dans le plasma.
Choo et ses collaborateurs ont donc essayé de détecter directement le génome du virus [8]. Ils ont développé une bibliothèque d’ADN complémentaire du matériel infectieux, construite avec des amorces aléatoires, à partir de sérums de patients atteints de cette hépatite non-A, non-B. Un de ces clones d’ADN a été identifié comme codant un antigène associé au virus. Il semblait être complémentaire d’un brin d’ARN issu du génome du pathogène, rapprochant ce dernier des Flaviviridae et des Togaviridae. Suite à ce travail novateur, le virus « de l’hépatite non-A, non-B » fut renommé « virus de l’hépatite C ».
Pathogénicité et cycle viral
Le VHC peut enclencher l’apoptose, voire la nécrose et l’autophagie des hépatocytes [9]. L’espace occupé préalablement par les hépatocytes apoptotiques va alors être comblé par du tissu conjonctif produit par les fibroblastes, conduisant à la fibrose hépatique.
Cycle viral
Il est important de comprendre les étapes clefs du cycle viral du VHC pour appréhender le mode d’action des différents traitements et sa pathogénicité. Cependant, les mécanismes moléculaires qui sous-tendent ce cycle ne sont pas complètement élucidés et restent extrêmement complexes. Nous avons donc sélectionné quelques protéines importantes dans ce cycle afin de présenter les cibles des traitements et d’expliquer la relation particulière du VHC avec le foie.
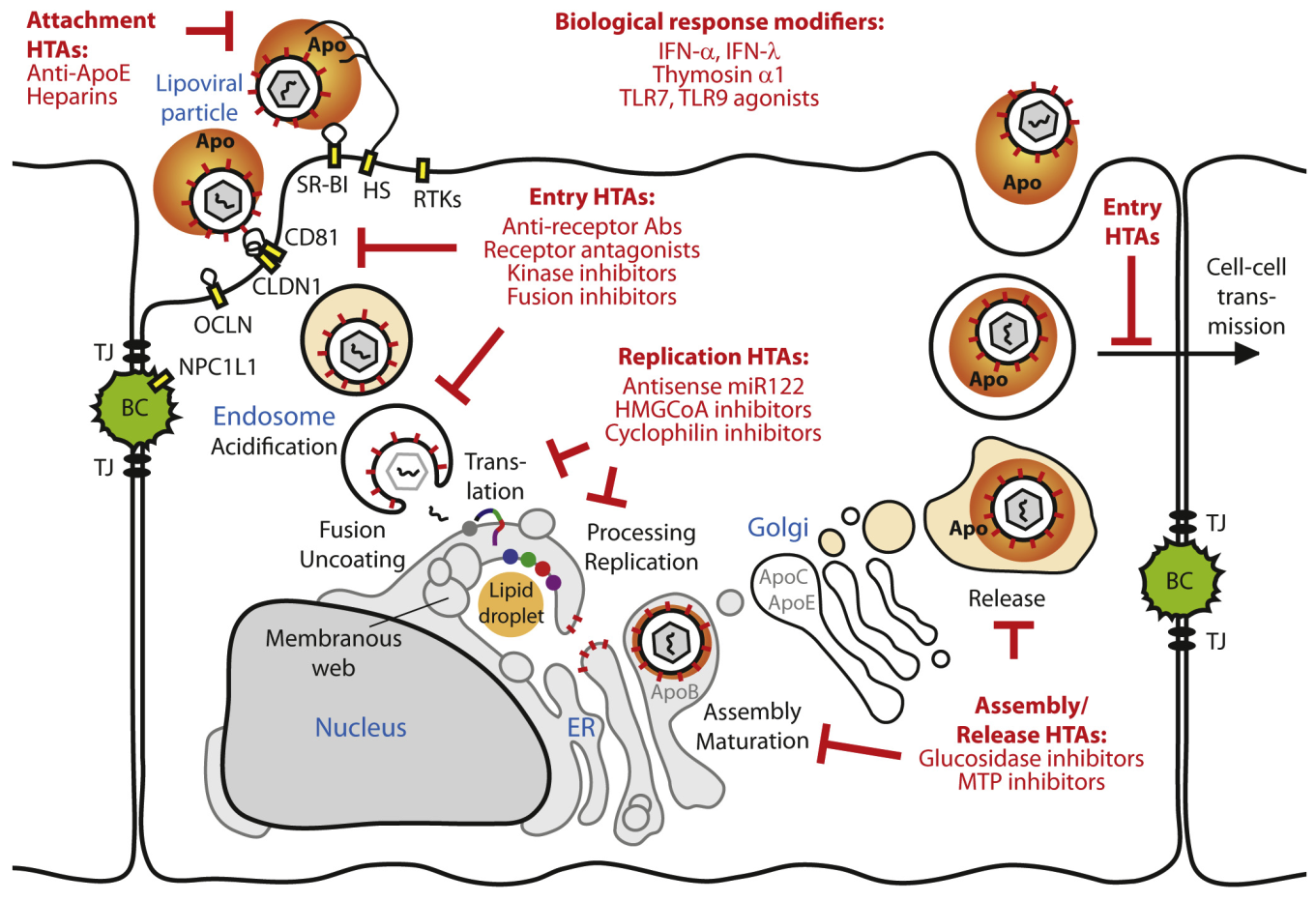
Le cycle viral du VHC présente des étapes clefs : son entrée dans l’hépatocyte, la traduction de ses protéines puis la mise en place de son complexe de réplication (grâce aux clivages activateurs réalisés par sa protéase NS3/4A), la réplication de son ADN (polymérase NS5B), l’assemblage de sa capside avec les gouttelettes lipidiques (lipid droplet), puis avec son ARN (en noir) et son enveloppe glycoprotéique (via l’action de NS5A). Les traitements utilisés pour lutter contre le VHC (en rouge) ciblent ces différentes étapes.
La première étape du cycle de réplication du VHC est son entrée dans la cellule, grâce à l’interaction de ses glycoprotéines de surface E1 et E2 avec la membrane basolatérale des hépatocytes, au contact du flux sanguin [11]. In vivo, l’entrée du VHC dans la cellule se fait grâce à une interaction séquentielle avec le récepteur aux lipoprotéines de faible densité (LDL), la tétraspanine CD81 (une protéine ubiquitaire) [12], la claudine 1 [13], l’occludine [14] et le récepteur scavenger de classe B, type 1 (SR-B1). Par la suite, le virus est endocyté dans des vésicules à clathrine, qui fusionnent avec les endosomes précoces. L’acidification de la vacuole permet la fusion de l’enveloppe du virus avec la membrane de l’endosome, conduisant au relargage de l’ARN viral dans le cytosol [11].
La persistance de l’ARN viral dans la cellule nécessite un micro-ARN spécifique du foie : miR122 [15]. Ce dernier recrute des protéines cellulaires qui vont se fixer en 5’ des ARN viraux, empêchant ainsi leur dégradation par les exonucléases de l’hôte [16].
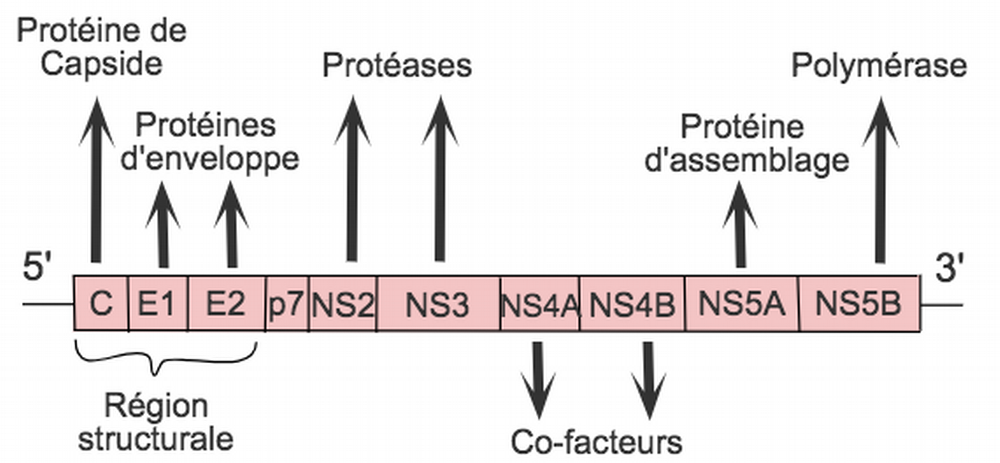
Le génome du VHC est composé de 9600 nucléotides.
L’ARN viral est tout d’abord traduit en une polyprotéine qui sera maturée par les protéines virales et cellulaires. Dix protéines matures sont produites. Il s’agit de protéines structurales : la protéine Core (impliquée dans la formation de la capside), les glycoprotéines d’enveloppe E1 et E2 ; ainsi que de protéines non structurales : p7, NS2, NS3, NS4A, NS4B, NS5A et NS5B [11]. L’ARN viral sert également de matrice pour être amplifié par l’ARN polymérase NS5B.
Après une accumulation des protéines structurales et de l’ARN viral dans le cytosol, la formation des virions (morphogenèse) peut commencer. Dans un premier temps, la protéine Core, formant la capside virale, va se lier aux gouttelettes lipidiques intracellulaires. La répartition de ces gouttelettes est totalement modifiée par la réplication du virus : alors qu’elles sont physiologiquement réparties équitablement dans tout le cytosol des hépatocytes, elles se retrouvent concentrées autour du noyau lors de la réplication du virus [17]. Il est important de noter que si l’on bloque certaines protéines nécessaires à la genèse des VLDL (lipoprotéines de très basse densité), telle que la MPT (protéine de transfert des triglycérides microsomaux), le virus ne peut plus se répliquer [18].
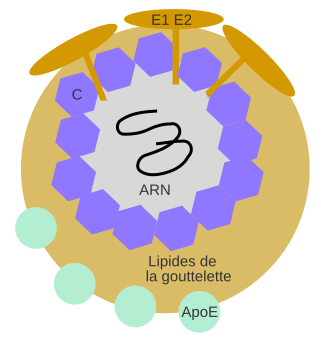
L’ARN du VHC se trouve dans une capside protéique, elle-même entourée de lipides issus des gouttelettes lipidiques cellulaires. Le VHC est un virus enveloppé, qui est en fait inclus dans une gouttelette lipidique. Sa couche externe est constituée d’un manteau de glycoprotéines virales (E1 et E2) et d’apolipoprotéines d’origines cellulaires (principalement ApoE).
D’après Virology and cell biology of the hepatitis C virus life cycle – An update , Dubuisson et coll., figure 3.
L’association lipide-capside entoure l’ARN fraîchement répliqué, de sorte que le virion se retrouve inclus dans la gouttelette lipidique, puis se lie avec les autres protéines structurales comme les hétérodimères de glycoprotéines d’enveloppe (E1 et E2) issus du réticulum endoplasmique. On retrouve également associées aux lipoviroparticules les apolipoprotéines A1, B, C1, C3 et E [19]. Seule l’apolipoprotéine E semble strictement nécessaire à l’obtention de virions fonctionnels [20]. La protéine virale NS5A joue également un rôle important dans l’assemblage du virus [21]. Même si son mécanisme d’action précis et toutes ses cibles ne sont pas encore totalement identifiés, son absence empêche totalement la formation de nouveaux virions.
Les virions ainsi formés seront ensuite exocytés et pourront infecter de nouvelles cellules.
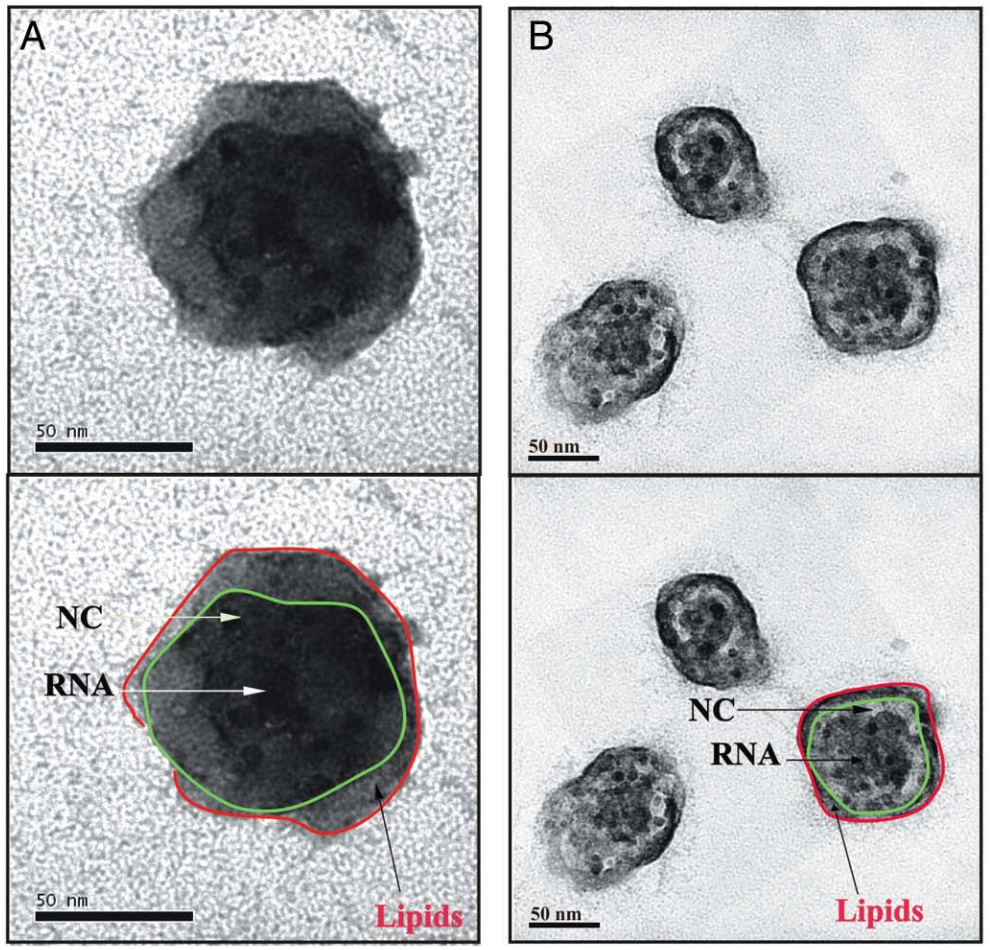
L’ARN du virus (RNA) est entouré par la nucléocapside (NC), elle-même incluse dans une couche lipidique (lipids). La colonne B met en évidence la taille variable des virions.
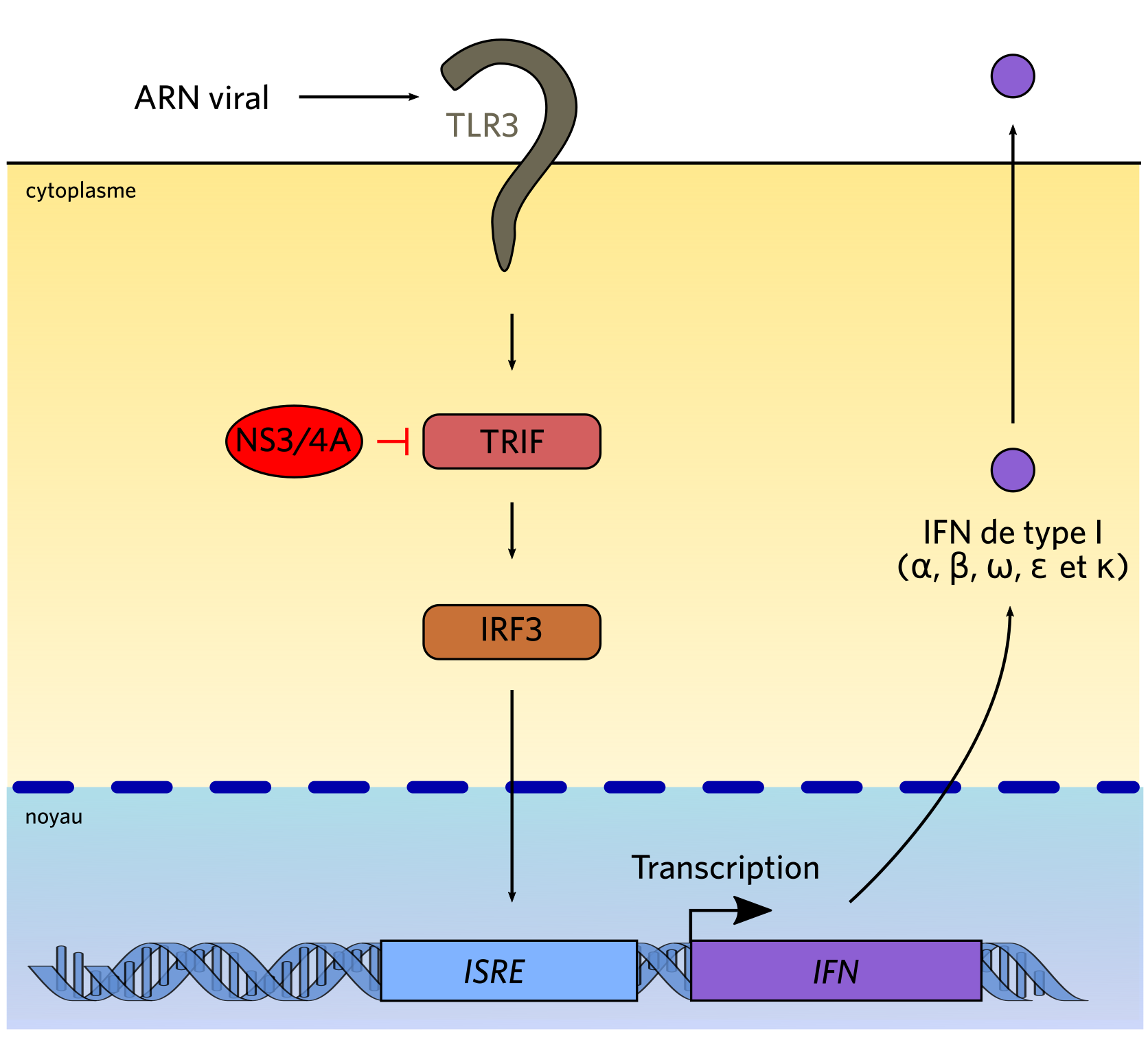
La protéine virale serait capable de cliver TRIF, protéine adaptatrice du TLR3.
Échappement au système immunitaire et pathogénicité
Le VHC présente de multiples techniques d’échappement au système immunitaire. Par exemple, il interfère avec la voie de production des interférons (IFN) de type I, molécules antivirales sécrétées par les cellules. La protéase NS3/4A est en effet capable de cliver des protéines clefs de la voie de synthèse des interférons, telles que TRIF [10], protéine adaptatrice du TLR3, ou IPS-1 [22] (figure 5). Par ailleurs, la protéine C du virus interagit avec la voie Stat, cascade de signalisation induite par l’IFN de type I endogène ou exogène [23]. Enfin, la protéine virale NS5A bloque quant à elle la protéine kinase R (PKR), activée également par l’IFN de type I et essentielle aux mécanismes de défenses de la cellule (diminution de la traduction) [24]. Le VHC peut par la suite épuiser le système immunitaire, causant des manifestations auto-immunes voire des lymphomes.
L’échappement du virus au système immunitaire est également à relier à son taux de mutations qui s’élève à 2.10-5 /nucléotide/réplication [25], du fait de son ARN polymérase peu fidèle. De plus le VHC génère une production très intense de virions (1012 nouveaux virions sont produits chaque jour chez un individu infecté), ce qui conduit à une variabilité génétique colossale. Un humain infecté abrite donc plusieurs quasi-espèces de virus, c’est-à-dire plusieurs populations de virions génétiquement très proches, mais présentant tout de même des différences de quelques nucléotides.
Premiers traitements
Les premiers traitements dans les années 90 étaient composés d’interféron-α(IFNα) recombinant, molécule antivirale naturellement produite par les cellules immunitaires comme les cellules dendritiques [26]. L’IFNα, injecté en sous-cutané, diminuait la réplication virale de manière non spécifique en activant les cellules NK et les lymphocytes T CD8, des cellules cytotoxiques détruisant les cellules infectées.
À la fin des années 90, l’IFNα a été associé à la ribavirine. Cet analogue nucléotidique de la guanosine remplace cette dernière lors de la réplication virale et empêche les autres bases azotées de venir se placer à sa suite [27]. Elle est capable de bloquer la réplication d’un certain nombre de virus, tels que le virus respiratoire syncitial (acteur majeur des infections virales pulmonaires basses durant l’enfance) ou des virus causant des fièvres hémorragiques comme le virus de Lassa. Au début des années 2000, l’IFNα a été pégylé (conjugué avec des chaînes de polyéthylène glycol) afin d’allonger sa demi-vie.
Cette combinaison ribavirine + Peg-IFNα montrait un succès mitigé : 40 à 50 % de succès contre le génotype 1 du VHC, jusqu’à 80 % contre les génotypes 2 et 3. L’efficacité du traitement était réduite chez les patients cirrhotiques [28].
L’ère des antiviraux directs
Trente ans seulement après la découverte du VHC, il est désormais possible de guérir complètement d’un point de vue virologique les patients atteints d’hépatite C. L’infection en tant que telle est résolue (il n’y a plus de virus dans l’organisme). Cependant les nombreuses complications et séquelles qu’elle a pu causer (cirrhose, hépatocarcinome…) ne régressent pas sous traitement et peuvent, dans les cas les plus graves, nécessiter une greffe de foie.
Les inhibiteurs de la protéase NS3/4A (télaprévir, boceprévir, glecaprévir, voxilaprévir)
La protéine non structurale 3 (NS3) est une protéase qui clive la polyprotéine initiale (issue de la traduction de tout le génome du VHC) au niveau des jonctions entre les différentes protéines virales non structurales (NS4B-NS5A, NS5A-NS5B…)[29]. NS4A est un cofacteur de NS3, modulant son activité enzymatique et sa localisation.
Les inhibiteurs de NS3/4A se lient de manière réversible au site actif de cette protéase, l’empêchant ainsi de cliver ses cibles [30, 31]. Ils ont été commercialisés en 2011, en association avec le Peg-IFNα et la ribavirine. Ces trithérapies augmentaient l’efficacité du traitement de manière significative et réduisait de moitié leur durée, mais au prix de nombreux effets secondaires et d’interactions médicamenteuses [32].
De nouvelles molécules, très efficaces et bien tolérées, appartenant également aux antiviraux directs, ont été commercialisées autour de 2014. Ces traitements permettent de s’affranchir de l’interféron, responsable de nombreux effets secondaires. Le taux de guérison en moins de 6 mois de traitement bien suivi, quels que soient les génotypes du VHC, est supérieur à 90 % [33].
Les inhibiteurs de la polymérase NS5B (sofosbuvir)
La protéine non structurale 5B produit l’ARN simple brin qui compose le génome du VHC [34]. Les inhibiteurs nucléotidiques de cette polymérase, comme le sofosbuvir, empêchent la réplication du virus car, une fois incorporés à l’ARN, ils en bloquent la polymérisation [35]. Le sofosbuvir, pris à raison d’un comprimé par jour et en association avec un autre antiviral, est efficace contre tous les génotypes du VHC. Il présente peu d’effets secondaires et peu d’interactions médicamenteuses (excepté avec les inducteurs enzymatiques).
Les inhibiteurs de la protéine NS5A (ledipasvir, pibrentasvir, velpatasvir)
La protéine non structurale 5A joue un rôle majeur dans l’assemblage du virus, dans l’encapsidation de son ARN et dans la formation de son complexe de réplication. Le mécanisme des molécules inhibitrices n’est pas encore complètement élucidé, mais elles agissent rapidement puisque l’on observe en moins de six heures une diminution de plus de deux ordres de grandeur de la réplication virale. Étonnement, si l’on place la protéine et ses inhibiteurs seuls dans un milieu, NS5A est toujours fonctionnelle, suggérant que les drogues agiraient sur des facteurs additionnels viraux ou cellulaires [36]. Il pourrait s’agir de certaines kinases cellulaires jouant un rôle dans la phosphorylation de NS5A. Les molécules thérapeutiques semblent également changer la distribution cellulaire de la protéine, en l’éloignant du réticulum endoplasmique et empêchant ainsi l’assemblage correct du virion [37]. Elles paraissent également limiter la formation des nouveaux complexes de réplication du virus. Ces molécules présentent peu d’effets secondaires et peu d’interactions médicamenteuses, tout en agissant efficacement à plusieurs étapes du cycle viral.
Le traitement actuel de l’hépatite C se compose d’une association de ces différents types d’antiviraux directs, en fonction du génotype et de l’avancement de la maladie, et permet une guérison totale de l’infection en 8 à 12 semaines de traitement [38].
Conclusion et perspectives
Le VHC semble être en bonne voie d’élimination, mais l’on ne peut pas encore évoquer son éradication : même si l’OMS envisage le traitement de 80 % des gens infectés d’ici 2030, le chemin à parcourir reste long.
Les tentatives de vaccins se sont pour le moment révélées infructueuses chez l’espèce humaine. Les protéines recombinantes de l’enveloppe glycoprotéique, E1 et E2, sont des facteurs immunogènes efficaces chez le chimpanzé, mais pas totalement chez l’humain [39]. Un vaccin trivalent (E2 solubles des trois génotypes principaux) a réussi à provoquer la production d’anticorps neutralisants contre tous les génotypes chez la souris et le singe rhésus [40]. De nombreuses équipes cherchent à faire exprimer à des virus non pathogènes pour l’humain (vaccine, variole du canari…), tout ou partie du génome du VHC (par exemple la protéine NS3). Certains de ces essais sont concluants chez la souris mais restent au stade préclinique [41]. Un vaccin basé sur les propriétés de la protéine S du virus de l’hépatite B est cours d’élaboration par l’équipe du professeur Roingeard et doit permettre d’obtenir un vaccin divalent capable d’induire des anticorps largement neutralisants contre les virus des hépatites B et C [42].
Un autre moyen de lutte contre l’hépatite C est le dépistage, car de nombreuses personnes sont porteuses du VHC sans en avoir connaissance. L’infection étant le plus souvent présente sous sa forme chronique et asymptomatique, avant que les complications n’apparaissent, il est important de mettre en place des moyens diagnostiques rapides et peu coûteux dans le monde entier. L’OMS vise 90 % de patients diagnostiqués d’ici 2030. Aujourd’hui l’outil de détection le plus efficace consiste à mesurer la charge virale (nombre de copies du génome du virus dans le sang), qui donne une bonne estimation de la réplication virale. La sérologie seule (présence d’anticorps anti-VHC) n’est pas suffisante, car elle peut témoigner d’une infection ancienne résolue.
Il est aussi nécessaire de continuer à suivre les patients, certes guéris de l’infection, mais dont les dommages hépatiques, comme une fibrose trop importante, augmentent significativement le risque de cancer. Il est donc également essentiel de déployer des moyens peu invasifs d’évaluation de la fibrose dans toutes les zones sensibles.
L’histoire de l’hépatite C est un vrai cas d’école et l’éradication de cette maladie dans un futur plus ou moins proche, comme cela a été le cas pour la variole au XXe siècle, ne semble pas impossible.
Bibliographie
- S. Navas, J. Martín, J. A. Quiroga, I. Castillo, et V. Carreño, « Genetic diversity and tissue compartmentalization of the hepatitis C virus genome in blood mononuclear cells, liver, and serum from chronic hepatitis C patients », J. Virol., vol. 72, no 2, p. 1640‑1646, févr. 1998.
- « Barriers of hepatitis C virus interspecies transmission. - PubMed - NCBI ». [En ligne]. Disponible sur: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pubmed/23217617.
- C. Niederau et al.., « Prognosis of chronic hepatitis C: results of a large, prospective cohort study », Hepatol. Baltim. Md, vol. 28, no 6, p. 1687‑1695, déc. 1998.
- « WHO | Global hepatitis report, 2017 », WHO. [En ligne]. Disponible sur: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/.
- P. Simmonds, « The origin and evolution of hepatitis viruses in humans », J. Gen. Virol., vol. 82, no Pt 4, p. 693‑712, avr. 2001.
- E. Drucker, P. G. Alcabes, et P. A. Marx, « The injection century: massive unsterile injections and the emergence of human pathogens », Lancet Lond. Engl., vol. 358, no 9297, p. 1989‑1992, déc. 2001.
- G. Magiorkinis et al.., « The global spread of hepatitis C virus 1a and 1b: a phylodynamic and phylogeographic analysis », PLoS Med., vol. 6, no 12, p. e1000198, déc. 2009.
- Q. L. Choo, G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, et M. Houghton, « Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome », Science, vol. 244, no 4902, p. 359‑362, avr. 1989.
- O. V. Masalova et al.., « Modulation of Cell Death Pathways by Hepatitis C Virus Proteins in Huh7.5 Hepatoma Cells », Int. J. Mol. Sci., vol. 18, no 11, nov. 2017.
- K. Li et al.., « Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF », Proc. Natl. Acad. Sci. U. S. A., vol. 102, no 8, p. 2992‑2997, févr. 2005.
- J. Dubuisson et F.-L. Cosset, « Virology and cell biology of the hepatitis C virus life cycle: an update », J. Hepatol., vol. 61, n o 1 Suppl, p. S3‑S13, nov. 2014.
- P. Pileri et al.., « Binding of Hepatitis C Virus to CD81 », Science, vol. 282, no 5390, p. 938‑941, oct. 1998.
- « Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry | Nature ». [En ligne]. Disponible sur: https://www.nature.com/articles/nature05654.
- « Human occludin is a hepatitis C virus entry factor required for infection of mouse cells | Nature ». [En ligne]. Disponible sur: https://www.nature.com/articles/nature07684.
- « Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA | Science ». [En ligne]. Disponible sur: http://science.sciencemag.org/content/309/5740/1577.
- « Stabilization of hepatitis C virus RNA by an Ago2–miR-122 complex | PNAS ». [En ligne]. Disponible sur: https://www.pnas.org/content/109/3/941.
- « Hepatitis C Virus Core Protein Induces Lipid Droplet Redistribution in a Microtubule‐ and Dynein‐Dependent Manner - Boulant - 2008 - Traffic - Wiley Online Library ». [En ligne]. Disponible sur: https://onlinelibrary.wiley.com/doi/full/10.1111/j.1600-0854.2008.00767.x.
- H. Huang et al.., « Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins », Proc. Natl. Acad. Sci., vol. 104, no 14, p. 5848‑5853, avr. 2007.
- R. Bartenschlager, F. Penin, V. Lohmann, et P. André, « Assembly of infectious hepatitis C virus particles », Trends Microbiol., vol. 19, no 2, p. 95‑103, févr. 2011.
- « Apolipoprotein E Codetermines Tissue Tropism of Hepatitis C Virus and Is Crucial for Viral Cell-to-Cell Transmission by Contributing to a Postenvelopment Step of Assembly | Journal of Virology ». [En ligne]. Disponible sur: https://jvi.asm.org/content/88/3/1433.
- T. Masaki et al.., « Interaction of Hepatitis C Virus Nonstructural Protein 5A with Core Protein Is Critical for the Production of Infectious Virus Particles », J. Virol., vol. 82, no 16, p. 7964‑7976, août 2008.
- X.-D. Li, L. Sun, R. B. Seth, G. Pineda, et Z. J. Chen, « Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity », Proc. Natl. Acad. Sci., vol. 102, no 49, p. 17717‑17722, déc. 2005.
- W. Lin et al.., « Hepatitis C virus core protein blocks interferon signaling by interaction with the STAT1 SH2 domain », J. Virol., vol. 80, no 18, p. 9226‑9235, sept. 2006.
- J. Pflugheber et al.., « Regulation of PKR and IRF-1 during hepatitis C virus RNA replication », Proc. Natl. Acad. Sci. U. S. A., vol. 99, no 7, p. 4650‑4655, avr. 2002.
- R. M. Ribeiro et al.., « Quantifying the diversification of hepatitis C virus (HCV) during primary infection: estimates of the in vivo mutation rate », PLoS Pathog., vol. 8, no 8, p. e1002881, 2012.
- M. H. Heim, « 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end », Nat. Rev. Immunol., vol. 13, no 7, p. 535‑542, 2013.
- « Mechanisms of action of ribavirin against distinct viruses. - PubMed - NCBI ». [En ligne]. Disponible sur: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pubmed/16287208.
- M. V. Preciado et al.., « Hepatitis C virus molecular evolution: transmission, disease progression and antiviral therapy », World J. Gastroenterol., vol. 20, no 43, p. 15992‑16013, nov. 2014.
- M. P. Manns, G. R. Foster, J. K. Rockstroh, S. Zeuzem, F. Zoulim, et M. Houghton, « The way forward in HCV treatment--finding the right path », Nat. Rev. Drug Discov., vol. 6, no 12, p. 991‑1000, déc. 2007.
- R. B. Perni et al.., « Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3-4A serine protease », Antimicrob. Agents Chemother., vol. 50, no 3, p. 899‑909, mars 2006.
- B. A. Malcolm et al.., « SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells », Antimicrob. Agents Chemother., vol. 50, n o 3, p. 1013‑1020, mars 2006.
- C. Hézode et al.., « Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC) - NCT01514890 », J. Hepatol., vol. 59, no 3, p. 434‑441, sept. 2013.
- « Emerging therapies for the treatment of hepatitis C. - PubMed - NCBI ». [En ligne]. Disponible sur: https://www-ncbi-nlm-nih-gov.gate2.inist.fr/pubmed/24106239. [Consulté le: 09-févr-2019].
- M. Niepmann, « Hepatitis C Virus RNA Translation », in Hepatitis C Virus: From Molecular Virology to Antiviral Therapy, R. Bartenschlager, Éd. Berlin, Heidelberg: Springer Berlin Heidelberg, 2013, p. 143‑166.
- A. Fung, Z. Jin, N. Dyatkina, G. Wang, L. Beigelman, et J. Deval, « Efficiency of incorporation and chain termination determines the inhibition potency of 2’-modified nucleotide analogs against hepatitis C virus polymerase », Antimicrob. Agents Chemother., vol. 58, n o 7, p. 3636‑3645, juill. 2014.
- J. J. Kohler et al.., « Approaches to hepatitis C treatment and cure using NS5A inhibitors », Infect. Drug Resist., vol. 7, p. 41‑56, 2014.
- P. Targett-Adams et al.., « Small molecules targeting hepatitis C virus-encoded NS5A cause subcellular redistribution of their target: insights into compound modes of action », J. Virol., vol. 85, no 13, p. 6353‑6368, juill. 2011.
- « Hépatite C: traitement actuel - Revue Médicale Suisse ». [En ligne]. Disponible sur: https://www.revmed.ch/RMS/2015/RMS-N-471/Hepatite-C-traitement-actuel.
- M. Houghton et S. Abrignani, « Prospects for a vaccine against the hepatitis C virus », Nature, vol. 436, no 7053, p. 961‑966, août 2005.
- X. Wang et al.., « A trivalent HCV vaccine elicits broad and synergistic polyclonal antibody response in mice and rhesus monkey », Gut, vol. 68, no 1, p. 140‑149, janv. 2019.
- A. von Delft et al.., « The generation of a simian adenoviral vectored HCV vaccine encoding genetically conserved gene segments to target multiple HCV genotypes », Vaccine, vol. 36, no 2, p. 313‑321, janv. 2018.
- Beaumont E, Roingeard P. « Chimeric hepatitis B virus (HBV)/hepatitis C virus (HCV) subviral envelope particles induce efficient anti-HCV antibody production in animals pre-immunized with HBV vaccine. » Vaccine. 2015 Feb 18;33(8):973-6.