Démonstration de l'équation de Michaelis et Menten. Détermination graphique de $v_{max}$ et $K_{M}$.
Les enzymes michaeliennes
Les enzymes sont des catalyseurs biologiques, c’est-à-dire des molécules capables d’accélérer des réactions chimiques spécifiques et qui sont retrouvées dans leur état initial à l’issue de la réaction catalysée.
Il existe différents mécanismes enzymatiques selon l’enzyme considérée. Les enzymes dites michaeliennes fonctionnent selon le mode suivant : un substrat S se lie avec une enzyme E pour donner un intermédiaire ES, appelé complexe enzyme-substrat, puis cet intermédiaire se dissocie pour donner un produit P avec régénération de l’enzyme E. Précision importante : chaque site actif se comporte indépendamment des autres, qu’ils soient physiquement séparés (un site actif par molécule) ou non (plusieurs sites actifs par molécule).
Bien entendu, il existe bien d’autres mécanismes enzymatiques. Citons en particulier les enzymes allostériques qui possèdent nécessairement plus d’un site actif par molécule (au moins deux) et pour lesquelles les caractéristiques cinétiques d’un site actif vont varier en fonction de l’état des autres sites de la même molécule (liés ou non liés à un substrat).
Chaque mécanisme se traduit par des caractéristiques cinétiques spécifiques (évolution de la vitesse de catalyse au cours du temps). Pour faire une étude cinétique, il faut mesurer la vitesse instantanée de la réaction à différents moments en ayant choisi avec soin les conditions initiales. À partir des mesures on peut tracer des courbes représentant la cinétique de la réaction, ce qui permet de déterminer certaines valeurs caractéristiques.
L’équation de Michaelis-Menten est une expression mathématique décrivant les paramètres cinétiques d’une réaction chimique catalysée par une enzyme michaelienne.
Détermination de l’équation de Michaelis-Menten
Au début du XXe siècle, Michaelis et Menten ont proposé un mécanisme réactionnel pour la transformation d’un substrat S en un produit P par une enzyme E. Ce mécanisme comporte deux étapes et fait intervenir un intermédiaire réactionnel : le complexe enzyme-substrat, noté ES.
$$E+S \begin{array}{c}
k_{1}\\
\rightleftharpoons\\
k_{-1} \end{array} ES \begin{array}{c}
k_{2}\\
\rightleftharpoons\\
k_{-2} \end{array}E+P $$
Avec $k_{1}$, $k_{-1}$, $k_{2}$ et $k_{-2}$ sont les constantes de vitesse des différents actes élémentaires.
Rappel de cinétique chimique : les concentrations des espèces à l'instant $t$ sont notées $[E]$, $[ES]$, $[S]$ et $[P]$. Les concentrations initiales de ces espèces sont notées $[E]_{0}$, $[ES]_{0}$, $[S]_{0}$ et $[P]_{0}$.
La vitesse de réaction (caractérisée par la vitesse d’apparition du produit, soit $v = \frac{d[P]}{dt}$) varie avec le temps (voir graphique 1).

La vitesse de la réaction à un instant t s'obtient en mesurant la pente de la tangente à la courbe à cet instant t. En début de réaction, la vitesse de réaction est constante, on parle d’état quasi-stationnaire. Durant cette période la quantité de produit $P$ reste négligeable donc les conditions de Michaelis sont respectées. La vitesse de réaction déterminée sur cette période correspond donc à la vitesse initiale $v_{i}$. Puis, on constate un infléchissement de la courbe, qui traduit une diminution progressive de la vitesse de la réaction. En effet, le produit continuant à s’accumuler, la réaction inverse (disparition du produit) devient non négligeable. À plus long terme, la concentration en produit atteint un plateau, la quantité maximale de produit pouvant apparaître dépendant de la quantité de substrat introduite au début de l’expérience et des constantes cinétiques de la réaction. On constate que pour des concentrations de substrat croissantes, la concentration finale de produit ainsi que la vitesse initiale de réaction sont également croissantes.
En début de réaction, la vitesse de réaction est quasi-constante et égale à la vitesse initiale, notée $v_{i}$ ou $v_{0}$. En fin de réaction, la vitesse tend vers zéro. En effet, soit la réaction est totale et une fois le substrat épuisé il n’y a plus de réaction possible (donc $v = 0$), soit un équilibre s'instaure entre la formation de produit ($ES \rightarrow E + P$) et sa destruction ($E + P \rightarrow ES$) ce qui, là encore, se traduit par une vitesse de réaction nulle.
L’équation de Michaelis-Menten donne une expression de la vitesse initiale de réaction $v_{i}$ en fonction de grandeurs connues (fixées par l’expérimentateur ou mesurées).
Pour cela il faut se placer dans des conditions expérimentales particulières, à savoir : concentration en substrat $[S]$ très largement supérieure à la concentration totale en enzyme $[E]_{0}$ et absence ou quasi-absence de produit $P$. La première condition est obtenue en choisissant des quantités adaptées d’enzyme et de substrat à introduire dans le milieu réactionnel, la seconde en réalisant les mesures suffisamment rapidement pour que la quantité de substrat transformé en produit soit faible. On parle de conditions de Michaelis. À partir du moment où ces conditions sont réunies, on peut effectuer les approximations suivantes :
- La concentration en produit $[P]$ étant nulle ou faible, la vitesse d’apparition du complexe $ES$ par réaction entre $E$ et $P$ ($v_{-2} = k_{-2}[E][P]$) est négligeable devant $v_{1} = k_{1}[E][S]$.
- On se place dans le cadre de l'approximation des états quasi-stationnaires, ce qui revient à considérer que la concentration $[ES]$ reste constante. Mathématiquement, cela se traduit par $\frac{d[ES]}{dt} = 0$.
- $[S]_{0}$ étant très grand par rapport à $[E]_{0}$, la concentration maximale en complexe $[ES]$ est limitée par $[E]_{0}$ et sera donc toujours négligeable comparée à $[S]_{0}$, même à saturation de tous les sites actifs. Or $[S] = [S]_{0} - [ES]$, donc si $[ES]$ est négligeable face à $[S]_{0}$, il en découle l’approximation suivante : $[S] = [S]_{0}$.
Ces préalables étant posés, on peut commencer à faire un traitement mathématique simple de la cinétique de la réaction.
Étape 1
À tout instant, la vitesse de réaction $v$ est donnée par $v = \frac{d[P]}{dt} = k_{2}[ES] – k_{-2}[E][P]$.
Si l’on considère maintenant la vitesse initiale de réaction $v_{i}$, d’après l’approximation 1 on peut négliger le terme $k_{-2}[E][P]$, ce qui donne $v_{i} = k_{2}[ES]$.
$[ES]$ est une valeur qui n’est pas fixée par l’expérimentateur, et qui ne peut pas être mesurée (pas plus que $[E]$ et $[S]$, contrairement à $[E]_{0}$ et $[S]_{0}$). Pour avoir une expression utilisable, il faut donc trouver une expression de $[ES]$ qui utilise des valeurs qui peuvent être connues (qu’elles correspondent aux conditions expérimentales fixées par l’expérimentateur ou qu’elles puissent être mesurées).
Il est également nécessaire de faire la distinction en $v_{i}$ et $v_{max}$. Quelles que soient les conditions, $v_{i} = k_{2}[ES]$. Par contre, il existe une valeur particulière de $v_{i}$, la vitesse maximale $v_{max}$. Celle-ci est atteinte lorsque $[ES] = [E]_{0}$, c’est-à-dire lorsque tous les sites actifs sont saturés. La $v_{max}$ est donc la vitesse maximale de catalyse pour une concentration donnée d’enzyme. Elle est obtenue à saturation de l’enzyme, autrement dit lorsque tous les sites actifs de toutes les molécules d’enzyme sont occupés. Sa valeur dépend évidemment d’autres conditions expérimentales (température, pH, etc.) mais nous n’aborderons pas leur influence dans cet article.
Étape 2
La variation de la concentration en complexe enzyme-substrat ES est liée aux vitesses des différents actes élémentaires du mécanisme proposé par Michaelis et Menten. Certains contribuent à sa formation (actes élémentaires 1 et -2, tandis que d’autres contribuent à sa disparition (actes élémentaires -1 et 2). Ainsi :
$$\frac{d[ES]}{dt} = k_{1}[E][S] - k_{-1}[ES] - k_{2}[ES] + k_{-2}[E][P] = 0$$
Puisqu'on néglige $k_{-2}[E][P]$ et qu'on considère que $\frac{d[ES]}{dt} = 0$, on obtient :
$$\frac{d[ES]}{dt} = k_{1}[E][S] - (k_{-1}[ES] + k_{2}[ES]) = 0 \\ \Leftrightarrow k_{1}[E][S] - (k_{-1} + k_{2})[ES] = 0 \\ \Leftrightarrow \frac{[E][S]}{[ES]} = \frac{(k_{-1} + k_{2})}{k_{1}}$$
Pour simplifier on pose $K_{M} = \frac{(k_{-1} + k_{2})}{k_{1}}$. $K_{M}$ est appelée constante de Michaelis ; sa dimension est celle d’une concentration.
L’équation précédente devient donc $$\frac{[E][S]}{[ES]} = K_{M} \\ \Leftrightarrow [ES] = \frac{[E][S]}{K_{M}}$$
Étape 3
À tout instant, la concentration totale d’enzyme $[E]_{0}$ se répartit entre les molécules libres $E$ et les molécules complexées avec le substrat, $ES$ :
$$[E]_{0} = [E] + [ES] \Leftrightarrow [E] = [E]_{0} - [ES] $$
En remplaçant ce terme dans l’équation trouvée en fin d’étape 2 on obtient :
$$[ES] = \frac{([E]_{0} – [ES])[S]}{K_{M}} \\ \Leftrightarrow [ES] = \frac{[E]_{0}[S]}{K_{M}} - \frac{[ES][S]}{K_{M}} \\ \Leftrightarrow [ES] + \frac{[ES][S]}{K_{M}} = \frac{[E]_{0}[S]}{K_{M}} \\ \Leftrightarrow [ES] \bigg( 1 + \frac{[S]}{K_{M}} \bigg) = \frac{[E]_{0}[S]}{K_{M}} \\ \Leftrightarrow [ES] = \frac{\frac{[E]_{0}[S]}{K_{M}}}{1 + \frac{[S]}{K_{M}}} \\ \Leftrightarrow [ES] = \frac{\frac{[E]_{0}[S]}{K_{M}}}{\frac{K_{M} + [S]}{K_{M}}} \\ \Leftrightarrow [ES] = \frac{[E]_{0}[S]}{K_{M}} \times \frac{K_{M}}{K_{M} + [S]} \\ \Leftrightarrow [ES] = \frac{[E]_{0}[S]}{K_{M} + [S]}$$
Étape 4
D'après l'étape 1, $v_{i} = k_{2}[ES]$, et d'après l'étape 3, $[ES] = \frac{[E]_{0}[S]}{K_{M} + [S]}$, donc $$ v_{i} = k_{2} \frac{[E]_{0}[S]}{K_{M} + [S]}$$
Or, d’après le raisonnement développé à la fin de l’étape 1, $k_{2} [E]_{0} = v_{max}$.
On en déduit donc l’équation de Michaelis et Menten : $$ v_{i} = \frac{v_{max}[S]}{K_{M} + [S]}$$
À partir de cette équation, on peut déterminer la $v_{i}$ pour toutes les conditions initiales connues puisque $v_{max}$ et $K_{M}$ sont des constantes qui peuvent être déterminées expérimentalement (voir paragraphe suivant). $v_{i}$ ne dépend donc que de la concentration en substrat $[S]$, valeur connue puisqu’il s’agit de la concentration en substrat introduite par l’expérimentateur (rappelons que selon l’approximation 3, $[S] = [S]_{0}$).
Note : en fait, cette équation est celle de Michaelis-Menten modifiée par Briggs et Haldane. Dans la version initiale de Michaelis et Menten, le terme $k_{2}$ était négligé. En effet, la dissociation du complexe $ES$ en $E + S$ est beaucoup plus rapide que la réalisation de la réaction catalysée $ES \rightarrow E + P$, ce qui revient à dire que la constante $k_{2}$ est très faible comparée à la constante $k_{-1}$. De ce fait, la constante $K_{S} = \frac{k_{-1}}{k_{1}}$ (correspondant à la constante de dissociation de $\frac{[E][S]}{[ES]}$) était utilisée en lieu et place de $K_{M}$.
La détermination graphique de la constante de Michaelis et de $v_{max}$
Pour déterminer les constantes $K_{M}$ et $v_{max}$, il faut faire une étude cinétique.
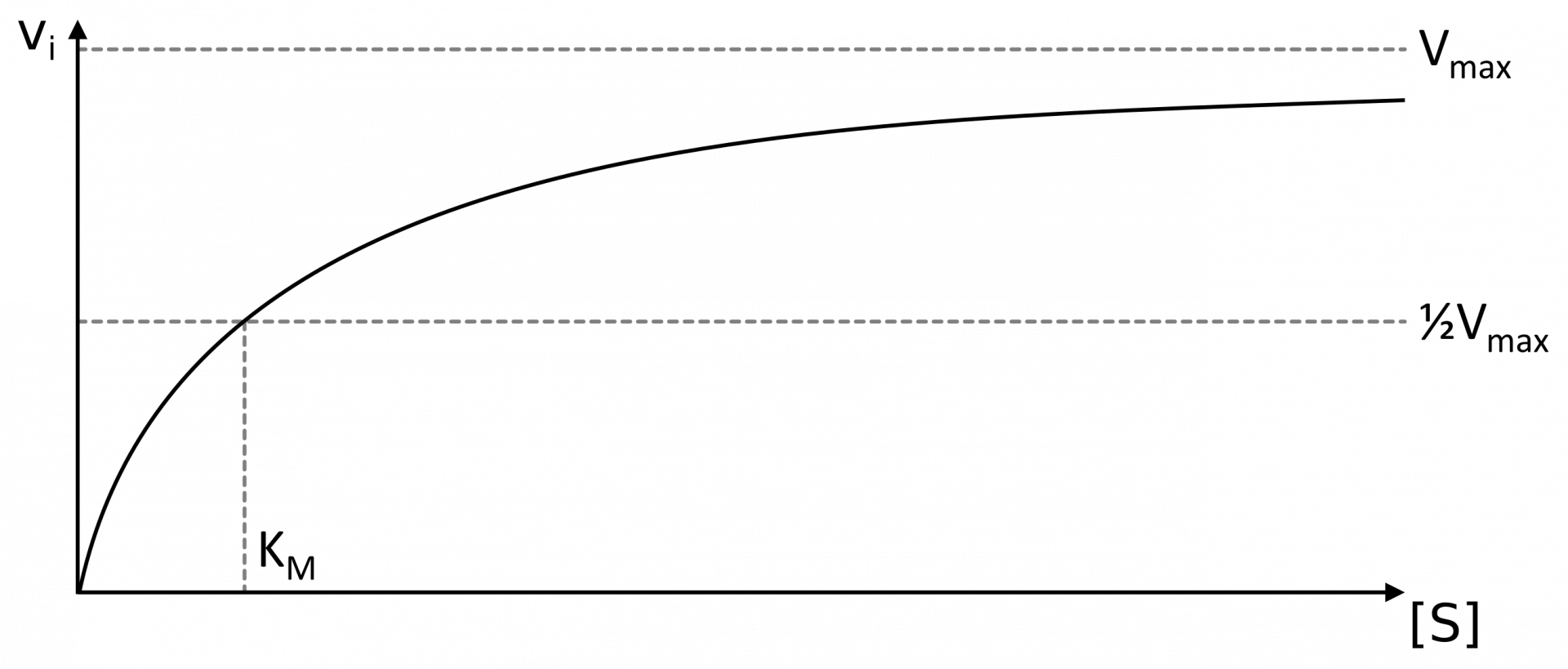
La détermination graphique de l’asymptote, et donc de $K_{M}$ et $v_{max}$, est peu précise.
Une première méthode consiste à tracer le graphique représentant les $v_{i}$ en fonction de la concentration en substrat $[S]$ utilisé. D’après l’équation de Michaelis, on peut en effet déduire que $v_{i}$ se rapproche asymptotiquement de $v_{max}$ lorsque $[S]$ augmente. Par ailleurs, lorsque $[S] = K_{M}$, $v_{i} = \frac{v_{max}}{2}$. On peut donc déterminer graphiquement $v_{max}$, puis $K_{M}$ (voir graphique 2). La difficulté vient du fait que la détermination graphique d’une asymptote est une nécessairement imprécise, entachant d’erreur la détermination de $K_{M}$ et $v_{max}$.
Pour améliorer la précision de la détermination graphique de ces deux constantes, il existe des représentations graphiques qui linéarisent les résultats, permettant des extrapolations plus précises.

Chaque point correspond à une mesure de $v_{i}$ pour une concentration en substrat $[S]$ donnée. Les intersections avec les axes permettent de déterminer graphiquement $K_{M}$ et $v_{max}$.
La méthode la plus connue est celle de Lineweaver et Burk dans laquelle on représente $\frac{1}{v_{i}} = f(\frac{1}{[S]})$ (voir figure 3).
$$ v_{i} = \frac{v_{max}[S]}{K_{M} + [S]} \\ \Leftrightarrow \frac{1}{v_{i}} = \frac{K_{M} + [S]}{v_{max}[S]} \\ \Leftrightarrow \frac{1}{v_{i}} = \frac{K_{M}}{v_{max}[S]} + \frac{[S]}{v_{max}[S]} \\ \Leftrightarrow \frac{1}{v_{i}} = \frac{K_{M}}{v_{max}} \frac{1}{[S]} + \frac{1}{v_{max}} \\ $$
On reconnaît une équation de droite de type $ \frac{1}{v_{i}} = a \frac{1}{[S]} + b$, avec : $a = \frac{K_{M}}{v_{max}}$ (ce qui correspond à la pente de la droite) et $b = \frac{1}{v_{max}}$ (ce qui correspond à l’ordonnée à l’origine).
Cette représentation permet donc, à partir des mêmes données que précédemment, de déterminer graphiquement les valeurs de $v_{max}$ et de $K_{M}$ plus précisément qu’avec la représentation précédente.
Il existe bien entendu d’autres traitements mathématiques permettant d’améliorer la précision de la détermination de ces constantes et de nos jours les paramètres cinétiques sont déterminés par des méthodes de régression non-linéaires beaucoup plus efficaces mais qui sont moins accessibles.
Conclusion
S’il existe bien des enzymes dont le mécanisme n’obéit pas strictement au schéma très simple des enzymes michaeliennes, ce modèle permet d’obtenir au minimum une première approximation des paramètres cinétiques que sont $v_{max}$ et $K_{M}$.
Il faut aussi se rappeler que la mise au point de ce modèle et de l’équation associée, très simple à résoudre, a permis d’accéder à ces données à une époque où l’outil informatique n’existait pas. Il était alors beaucoup plus difficile d’approcher un résultat par de nombreux calculs.
De fait, il faudra attendre les années 1960 pour connaître une évolution conceptuelle aussi importante dans le domaine de l’enzymologie avec la découverte de l’allostérie.