

Après quelques rappels sur la notion de séquence nucléotidique, cet article définit en quoi consiste le séquençage d'un génome et en présente les modalités et contraintes techniques (ainsi que les aspects statistiques). Il fait le point sur les avancées du séquençage dans la biodiversité du vivant et termine en présentant les différents débouchés apportés par la connaissance du génome complet d'un individu ou d'une espèce.
Introduction
De très nombreux génomes, dont celui de l’être humain, sont aujourd’hui séquencés, et des génomes plus nombreux encore sont en voie de l’être. Un grand nombre de laboratoires, aussi bien publics que privés, participent (parfois de façon concurrente) à cet énorme effort qui révolutionne tant la biologie fondamentale que les biotechnologies.
Le but de ce document est de fournir quelques données sur les séquençages de génomes et leurs modalités de réalisation. Pour plus d’information, voir également le document le séquençage d’un ADN.
Les génomes séquencés
Rappel sur les séquences nucléotidiques
Les nucléotides, maillons élémentaires de l’ADN, peuvent être de 4 types différents dans cette molécule. Ils sont constitués d’une partie constante (squelette sucre-phosphate) et d’une partie variable, une base, du point de vue chimique. Les 4 bases présentes dans l’ADN sont notées A, T, G et C (Adénine, Thymine, Guanine et Cytosine).
La succession des bases le long d’un brin d’ADN est la séquence de ce brin. On parle alors de séquence nucléotidique (figure ci-dessous).
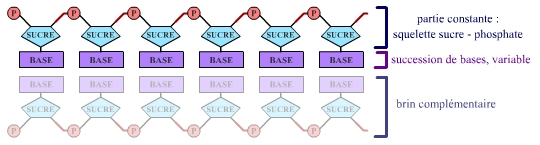
On peut donc exprimer la taille d’une séquence en nombre de bases – kilobases (kb) pour milliers de bases, mégabases (Mb) pour millions de bases, gigabases (Gb) pour milliards de bases – et la taille d’une molécule d’ADN en nucléotides, ou en paires de bases, pour se souvenir qu’une molécule d’ADN est formée de deux brins antiparallèles complémentaires.
Qu’est-ce que le séquençage d’un génome ?
Le séquençage d’un génome consiste en la détermination de la séquence nucléotidique de l’ADN présent dans chaque cellule d’un organisme donné.
Cette détermination est en général d’autant plus difficile que le génome étudié est grand et riche en séquences répétées.
Les virus, qui possèdent de petits génomes dénués de séquences répétées (entre 3 000 et 150 000 paires de bases, souvent moins de 10 000), ont ainsi été les premiers « organismes » séquencés, et représentent aujourd’hui encore la majorité d’entre eux.
La première bactérie a été séquencée en 1995, et de nombreux autres procaryotes ont, depuis, été séquencés intégralement. La taille de leur génome est de l’ordre de quelques millions de paires de bases (mégabases, Mb). La difficulté est tout autre pour les organismes eucaryotes : la grande taille de leur génome (2 à 3 milliards de paires de bases pour les mammifères, par exemple) nécessite un travail cartographique préalable et, souvent, un effort concerté de plusieurs centres de séquençage. Toutefois, les « petits » génomes eucaryotes (tel celui de la paramécie, qui ne mesure « que » 100 Mb) peuvent aujourd’hui être séquencés sans cartographie préalable par un seul grand centre.
Si les eucaryotes intégralement séquencés demeurent moins nombreux que les procaryotes et virus, leur nombre est en constante augmentation. Certaines séquences sont à l’état d’ébauches très fragmentaires et incomplètes ; d’autres donnent une vision beaucoup plus complète du génome. Cela reflète l’effort de séquençage consenti ainsi que la stratégie adoptée par les auteurs du séquençage, comme on le verra plus loin. Au mois de mars 2014, on comptait 12 919 génomes cellulaires complètement séquencés (hors virus), 27 399 en cours de séquençage et 996 en projet.
Pour ce qui est de l’Homme, la première séquence du génome humain, annoncée à grands cris médiatiques fin 2000, n’était en rien une séquence complète. Celle-ci est disponible depuis avril 2003, à quelques « trous » près.
La liste qui suit donne quelques exemples de génomes séquencés parmi les premiers qui le furent.
-
Virus : 3 778 virus séquencés au 4 mars 2014, dont le VIH (virus d’immunodéficience humaine).
-
Procaryotes :
-
Archaebactéries : 319 génomes entièrement séquencés et 447 partiellement, au 4 mars 2014.
-
Eubactéries : 12 286 génomes entièrement séquencés et 20 403 partiellement, au 4 mars 2014. Exemples :
-
Escherichia coli
-
Agobacterium tumefaciens
-
Haemophilus influenzae Rd. ( premier génome cellulaire séquencé, en 1995 )
-
-
-
Eucaryotes : 314 génomes entièrement séquencés et 6660 partiellement, au 4 mars 2014. Exemples (les 5 premiers cités sont les 5 premiers publiés, avec la réserve citée pour celui de l’Homme) :
-
Saccharomyces cerevisiae (levure, premier eucaryote à être séquencé en 1997, plusieurs souches séquencées de nos jours)
-
Caenhorabditis elegans (ver nématode)
-
Drosophila melanogaster (mouche du vinaigre)
-
Arabidopsis thaliana (arabette, petite plante de la famille du chou)
-
Homo sapiens (nous, l’espèce humaine : plusieurs individus séquencés dont Watson)
-
Neurospora crassa ( champignon ascomycète )
-
Anopheles gambiae (moustique)
-
Takifugu rubripes (fugu, poisson-ballon consommé au japon)
-
Mus musculus (souris)
-
Plasmodium falciparum (parasite intracellulaire responsable du paludisme)
-
Oriza sativa (riz : deux sous-espèces séquencées ; japonica et indica)
-
De nombreux projets de séquencage sont actuellement en cours de réalisation, ou à l’étude. Ces projets se comptant par milliers, il serait trop long de tous les énumérer ici…
Pour plus d’informations à ce sujet le site GOLD (Genome OnLine Database) qui recense tous les génomes séquencés, ainsi que les projets en cours.
Le séquençage d’un génome : comment ça marche ?
Depuis la fin des années 1970 et l’avènement des techniques de la biologie moléculaire, il est possible de séquencer un brin d’ADN, c’est-à-dire de lire l’enchaînement, ou séquence, des nucléotides constitutifs de cette molécule. Cela se ramène en fait à déterminer la succession des bases, la seule partie variable des nucléotides (pour plus d’informations sur les principes de la lecture de l’ADN, voir le document « Le séquençage d’un ADN »). Cependant, les techniques actuelles ne permettent de lire, à chaque opération de séquençage, qu’un millier de bases au plus. Or la partie « séquençable » du génome humain comprend 2,9 milliards de paires de bases (gigabases, Gb) ! Il est donc impossible de lire l’ensemble d’un génome en une fois. Il est, de toute façon, impossible de manipuler des molécules d’ADN de plusieurs dizaines, voire centaines de millions de bases (l’ordre de grandeur de celles qui constituent les chromosomes humains).
Remarque
On parle de « partie séquençable » du génome humain (2,9 Gb, pour un total de 3,2 Gb). En effet, il n’est pas possible, techniquement, de déterminer la séquence de certaines régions presque exclusivement constituées de séquences répétées, telles que les centromères, les télomères ou les bras courts de certains chromosomes. Il y a deux raisons à cela : d’une part, il est difficile d’isoler des fragments d’ADN de taille convenable issus de ces régions ; d’autre part, il n’est pas possible de reconstituer la séquence complète à partir de morceaux de séquences pratiquement identiques.
De ce fait, seule la séquence de la partie dite euchromatique du génome peut être effectivement déterminée.
Dans la suite, quand seront indiqués des pourcentages de l’ADN humain séquencé, c’est toujours à ces 2,9 milliards de paires de base que l’on se référera, et non aux 3,2 milliards du génome dans son entier.
Le principe de base, dans tout séquençage d’un génome, consiste donc à fragmenter de façon aléatoire ce génome – ou de grands morceaux d’ADN dérivés du génome – pour obtenir des morceaux d’ADN de quelques milliers de paires de bases, faciles à manipuler. Les extrémités d’un grand nombre de ces petits fragments sont alors séquencées. La séquence complète du génome – ou du grand morceau de génome – est ensuite reconstruite à partir de ces séquences unitaires, ou lectures, sur la base des chevauchements entre les séquences (si les séquences sont chevauchantes, c’est que les fragments d’ADN dont elles dérivent ont une partie de leur longueur en commun ; la cassure étant aléatoire, les molécules d’ADN de l’échantillon ne sont pas toutes cassées aux mêmes endroits).
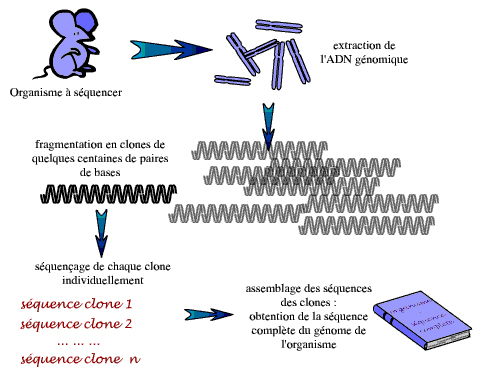
Cette méthode présente toutefois certaines difficultés : tout d’abord, pour obtenir suffisamment de séquences chevauchantes et pour réduire au maximum les erreurs de séquençage, il faut atteindre un certain niveau de redondance, c’est-à-dire produire une quantité de séquences aléatoires représentant plusieurs fois la longueur de la séquence d’intérêt. Ceci conduit à un nombre très important de séquences à réaliser… Dans nombre de projets de séquençage, on détermine ainsi la séquence de 10 fois plus d’ADN que n’en comporte le génome étudié : on parle d’une profondeur de 10X. Dans ce cas de figure, chaque base de la séquence cible a été lue 10 fois en moyenne, mais certaines l’ont été davantage, d’autres moins et d’autres encore pas du tout. Même à 10X, des « trous » peuvent donc subsister, laissant la séquence finale très légèrement incomplète.
Le séquençage, compléments: fragments séquencés et lois statistiques
Dans un projet de séquençage, le rapport entre la longueur de l’ensemble des séquences lues mises bout à bout et la longueur du génome cible est nommé profondeur. Par exemple, si l’on séquence 25 millions de bases (Mb) pour un génome de 5 Mb, on a une profondeur de 5 équivalents génome, ce que l’on note 5X. Plus la profondeur est importante, plus nombreuses seront les lectures chevauchantes que l’on pourra assembler, et plus grande sera la fraction du génome couverte. Ceci permet d’obtenir une séquence finale la plus complète possible, avec un minimum de “trous”, régions non séquencées. Toutefois, si l’augmentation de la profondeur du séquençage permet de diminuer ces lacunes de séquence, il arrive un seuil où il est plus économique de boucher les quelques trous restants de façon ciblée. Par ailleurs, il peut y avoir des biais de représentations qui font que certaines régions sont moins couvertes, voire pas du tout.
Il est possible de donner une représentation mathématique de ces trous dans la séquence finale. Cela demeure toutefois idéalisé et l’obtention d’une couverture donnée nécessite en pratique une profondeur souvent supérieure à la profondeur suffisante en théorie. C’est particulièrement vrai dans le cas de grands génomes comme celui de l’homme.
Si la longueur moyenne n des lectures est très petite devant la longueur L de la séquence cible et que le nombre de lectures est très grand, alors on peut considérer que la probabilité qu’une base de la séquence cible soit représentée dans x lectures suit une loi de Poisson, Pxe -P / x ! où P est la profondeur. Voyons alors le résultat de l’assemblage pour deux valeurs de la profondeur, en choisissant comme séquence cible le génome humain.
|
Exemple 1 |
Exemple 2 |
|
|
Soit L la taille de l’ADN étudié. |
L = 3.109 nucléotides |
L = 3.109 nucléotides |
|
On a donc la profondeur de lecture : profondeur P = N / L |
P = (3.1010) / (3.109) = 10 X |
P = (9.109) / (3.109) = 3 X |
|
Soit n la taille de chaque lecture. |
n = 1000 nucléotides |
n = 1000 nucléotides |
|
Alors on a : |
||
|
Fraction d’ADN représenté par au moins une lecture : |
||
|
ADN lu = 1 – e -N/L |
ADN lu = 1 – e-10 = 0,9999546 |
ADN lu = 1 – e-3 = |
|
Nombre de trous : |
||
|
N_trous = (N/n).e -N/L |
N_trous = |
N_trous = |
|
Taille moyenne des trous : |
||
|
T_trous = L.n/N |
T_trous = |
T_trous = |
Ce calcul montre que pour un même génome de 3 milliards de paires de bases, on obtient une couverture finale très différente, selon qu’une profondeur de 3 ou de 10 est choisie pour le séquençage.
Ces parties de la séquence cible qui ne sont pas couvertes par les lectures effectuées au hasard constituent une seconde difficulté : du fait de ces trous, le résultat de l’assemblage des lectures chevauchantes ne donne pas une séquence continue, mais plusieurs blocs de séquence continue, ou « contigs », qu’il peut être difficile dans un premier temps d’orienter et d’ordonner les uns par rapport aux autres, et d’assigner à un emplacement dans le génome. Séquencer davantage améliore la situation, mais un travail ciblé peut être nécessaire pour combler certains trous.
Ces difficultés expliquent que les premiers génomes séquencés ont d’abord été des génomes de très petite taille : ceux de virus. Les progrès techniques (mise au point des séquenceurs automatiques, augmentation de la puissance des ordinateurs, algorithmes bio-informatiques pour l’assemblage des séquences…) ont ensuite rendu possible le séquençage de génomes de plus en plus grands : le premier génome bactérien ( Haemophilus influenzae) en 1995, puis le premier eucaryote entier (Saccharomyces cerevisiae) en 1996. La mise sur pied de grands centres de séquençage, l’afflux de fonds publics ou caritatifs et la réduction des coûts du séquençage, au cours de la décennie écoulée, ont permis de s’attaquer aux génomes des eucaryotes supérieurs, dont celui de l’Homme.
De façon schématique, deux stratégies de séquençage sont actuellement utilisées :
-
la stratégie du séquençage aléatoire global, « whole genome shotgun » ;
-
la stratégie « clone par clone », ou du « shotgun hiérarchique », qui suppose la construction préalable ou concomitante d’une carte physique. Cette dernière stratégie a notamment été utilisée par le consortium international en charge du séquençage du génome humain.
En fait, on voit se développer de plus en plus de stratégies « mixtes », comme celle utilisée par le consortium public responsable du séquençage du génome de la souris. L’opposition apparemment tranchée entre ces deux stratégies est de moins en moins nette. Pour plus de clarté, nous les expliquerons cependant séparément.
Stratégie du séquençage aléatoire global
Historique
Cette stratégie de séquençage « aléatoire » appliquée à l’ensemble du génome (ou « whole genome shotgun ») a été utilisée dès les débuts du séquençage, en 1982, pour venir à bout du génome du bactériophage lambda. C’est la stratégie retenue aujourd’hui pour tous les génomes bactériens. On l’associe plus volontiers aux sociétés privées, telles que Celera Genomics ou Syngenta, parce qu’elle est rapide et économique. Celera a ainsi accompli le séquençage de plusieurs organismes par une stratégie aléatoire globale. Toutefois, il faut remarquer que, pour de grands génomes comme celui de l’Homme, ces sociétés se sont souvent appuyées sur des données de cartographie établies par les chercheurs académiques. En outre, pour ces grands génomes, on tend aujourd’hui à utiliser des stratégies mixtes, mêlant séquençage aléatoire global et « clone par clone ».
Le séquençage du génome humain a donné lieu à une lutte médiatique entre Celera Genomics et le consortium international responsable du « projet Génome humain », qui utilisait quant à lui une stratégie « clone par clone ». Celera Genomics a prétendu qu’elle avait procédé essentiellement par séquençage aléatoire global, mais la réalité est plus compliquée (voir plus bas). En revanche, des génomes comme celui de l’anophèle, à l’état d’ébauche, ont bien été séquencés selon cette seule stratégie. Les données du projet de séquençage du génome de ce moustique seront fournies ci-dessous à titre d’exemple, mais nous allons d’abord exposer le principe du séquençage aléatoire global à partir d’un projet plus modeste, celui de la première bactérie séquencée : Haemophilus influenzae Rd.
La technique « shotgun » repose sur un principe simple : découper un génome en un grand nombre de fragments de petite taille. Les extrémités d’une partie de ces fragments sont ensuite séquencées, puis ces séquences sont assemblées sur la base de leurs chevauchements grâce à des programmes informatiques pour essayer de produire une séquence complète. Les difficultés d’une telle technique sont à deux niveaux : (1) avoir assez de fragments pour couvrir le génome dans son entier, et (2) réussir l’assemblage.
Combien de fragments pour un génome ?
Le génome d’Haemophilus influenzae Rd comporte 1,8 millions de paires de bases (Mb). Le centre de séquençage privé TIGR (« The Institute for Genomics Research » de Rockville, dans l’état du Maryland aux États-Unis) en a déterminé la séquence et l’a publiée en 1995 dans la revue Science. Comment les chercheurs de TIGR s’y sont-ils pris ? Pour commencer, ils ont cassé le génome de cette bactérie par un moyen mécanique et ont constitué une « banque » de petits fragments d’ADN d’environ 2000 paires de bases (pb). Ils ont ensuite séquencé près de 20 000 de ces fragments, à une extrémité ou aux deux, et ont retenu, en vue de l’assemblage, plus de 24 000 lectures (produits d’une opération unitaire de séquençage). D’une longueur moyenne de 470 paires de bases, ces lectures représentent plus de 11,6 Mb séquencés, soit 6,3 fois la longueur du génome d’H. influenzae (profondeur de 6,3X).
Pourquoi avoir séquencé autant ? Un raisonnement naïf nous fait conclure qu’il suffit de 4000 lectures environ pour couvrir tout le génome d’ H. influenzae (4000 x 470 pb = 1,88 Mb). Il n’en est rien ! En effet, l’ADN est extrait de nombreuses cellules (pour disposer de quantités suffisantes) et cassé de façon aléatoire ; chaque copie du génome est donc cassée en des points différents et les fragments obtenus sont chevauchants, comme nous l’avons déjà indiqué. En outre, l’échantillonnage des fragments en vue du séquençage se fait, là encore, au hasard (chaque lecture peut être assimilée à un tirage aléatoire d’un morceau de la séquence du génome). Par conséquent, si l’on prend juste le « bon » nombre de lectures (profondeur de 1X), un grand nombre d’entre elles couvriront les mêmes régions… L’ensemble du génome ne sera pas couvert.
Mais un ensemble de 4000 lectures couvrant parfaitement le génome sans aucun chevauchement, à supposer qu’on puisse l’obtenir, ne serait pas non plus d’une grande utilité. Comment alors ordonnerait-on ces séquences entre elles pour reconstituer le puzzle ? Le fait de disposer de fragments qui se chevauchent permet « d’assembler » les séquences lues à partir de ces fragments sur la base de leur similitude, et d’obtenir des blocs de séquence de plus grande taille. Cela permet aussi de corriger les éventuelles erreurs de séquençage grâce aux différentes lectures couvrant la même zone. Avec la quantité de fragments que l’on séquence, croissent donc : la longueur des blocs de séquence que l’on est en mesure d’assembler, la fraction du génome couverte, ainsi que la précision de la séquence.
Documents à télécharger
À partir d'un exemple très théorique, cette animation Flash (fichier swf) explique comment se réalise un séquençage. Une profondeur de 1X ne peut pas permettre d'obtenir une séquence complète. Il est ainsi nécessaire de séquencer bien plus d'ADN.
L’assemblage laisse des trous
Revenons à Haemophilus influenzae et voyons le résultat de l’assemblage de TIGR à 6X : la comparaison des 24 000 lectures entre elles (une opération gourmande en moyens informatiques) a permis d’assembler in fine 140 grands blocs de séquence continue, ou contigs (pour « contiguous sequence »). L’on pouvait s’attendre, sur la base d’un calcul statistique, à ce que les 140 trous correspondants soient petits : moins de 100 paires de bases en moyenne. Toutefois, on ne savait rien à ce stade de la position respective des contigs dans le génome : il restait à les ordonner et à les orienter.
Pour déterminer les relations de voisinage des contigs, les chercheurs de TIGR ont alors considéré les « liens clones », c’est-à-dire les lectures obtenues aux deux extrémités d’un même fragment d’ADN, et ont recherché parmi ces paires celles qui s’ancraient dans deux contigs différents. Cela permet de jeter un pont entre les deux contigs et de les orienter. En outre, le fragment d’ADN « à cheval » sur le trou entre les deux contigs peut faire l’objet d’un séquençage supplémentaire, ce qui permet de combler le trou.
Les chercheurs de TIGR ont réussi par ce moyen à assembler les 140 contigs en 42 échafaudages plus vastes, et à combler les 98 trous ainsi enjambés. Restait donc 42 trous « physiques » que n’enjambait aucun fragment d’ADN parmi ceux retenus pour le séquençage. Diverses méthodes ont été utilisées pour les combler, dont le séquençage des extrémités de fragments d’ADN de plus grande taille (20 000 paires de bases), susceptibles de fournir des liens clones sur une plus grande échelle. Cet effort de finition a livré la séquence complète du génome : les 1 830 137 paires de bases du chromosome circulaire d’Haemophilus influenzae Rd ont été déposées dans les bases de données et livrées à la curiosité des scientifiques du monde entier.
Documents à télécharger
Animation Flash (fichier swf)
Limites et intérêts de la stratégie du séquençage génomique aléatoire global
Le séquençage aléatoire global s’est aujourd’hui imposé dans le cas des génomes bactériens. Comment s’applique-t-il aux génomes plus grands et plus complexes ? L’exemple du séquençage du moustique africain Anopheles gambiae, le vecteur principal du paludisme, va permettre d’éclairer certaines limites de cette stratégie.
L’intérêt principal du séquençage aléatoire global réside dans le fait qu’il n’est pas nécessaire de réaliser une carte physique préalable, étape longue et fastidieuse, comme on le verra dans le chapitre suivant. Mais l’assemblage est rendu, de ce fait, particulièrement difficile : une très grande puissance de calcul est nécessaire, et le résultat n’est pas assuré… Un génome tel que celui de l’anophèle, long de 280 Mb (plus de 150 fois la taille du génome d’Haemophilus influenzae Rd), pose en effet de toutes autres difficultés que celui d’une bactérie : il est non seulement 100 fois plus long, mais aussi beaucoup plus riche en séquences répétées. Enfin, il est diploïde, et l’existence d’une hétérozygotie importante introduit des incertitudes et des erreurs lors de l’assemblage (les lectures assemblées dans la stratégie du séquençage aléatoire global peuvent en effet provenir de l’un ou l’autre des deux chromosomes homologues, ce qui n’est pas le cas, nous le verrons, dans la stratégie « clone par clone »). Comment déterminer alors, si deux séquences très similaires correspondent à des allèles d’un même locus (elles sont alors légitimement assemblées), ou à des séquences provenant de deux endroits différents du génome, issues d’une duplication récente et ayant peu divergé ?
Le projet de séquençage de l’anophèle, achevé en 2002, a été conduit principalement par Celera (90 % des lectures) et par le Genoscope. Plus de 4,5 millions de lectures ont été produites, pour la plupart appariées, ce qui représente une profondeur de 10X. Une différence avec l’exemple précédent est que les lectures proviennent du séquençage des extrémités de fragments d’ADN de tailles plus variées : à une majorité de petits fragments (2,5 et 10 kb) s’ajoutent cette fois une part importante de fragments de 50 kb, voire 100 kb, qui procurent des liens clones « à longue portée ». Grâce à de tels liens clones, les 19 000 contigs produits par l’assemblage ont pu être réunis en 9000 « échafaudages ». On voit donc qu’il s’agit d’une version très morcelée de la séquence du génome de l’anophèle, même si 90 % de la séquence est comprise dans 300 grands échafaudages. Si la finition d’une telle ébauche génomique était entreprise (rien n’est moins sûr !), son coût et sa difficulté seraient sans commune mesure avec ceux de la finition de la séquence d’H. influenzae. Pour un tel génome, et a fortiori pour celui d’un mammifère, la stratégie du séquençage aléatoire global est donc rapide et économique… si l’on compte s’en tenir à une ébauche de la séquence du génome. Cela convient à certains usages de la séquence, beaucoup moins à d’autres. En particulier, l’annotation précise des gènes nécessite une séquence de qualité « finie ».
Pour aller plus loin, et pour revenir sur certains aspects très médiatisés de ces séquençages génomiques, lire ci-dessous les commentaires sur la version de la séquence du génome humain proposée par Celera genomics.
Les difficultés évoquées au sujet du génome de l’anophèle sont plus épineuses encore dans le cas du génome d’un mammifère tel que l’homme, 10 fois plus grand et constitué à près de 50 % de séquences répétées (notons tout de même que le polymorphisme, chez l’homme, est moins important que chez l’anophèle). L’intérêt de la stratégie du séquençage aléatoire global, dans sa forme « pure », reste donc discuté pour de tels grands génomes. Les génomes de la souris et du rat ont d’ailleurs été récemment séquencés selon une stratégie « mixte », mêlant séquençage aléatoire global et effort cartographique.
Cette mise en garde pourra surprendre : certains lecteurs se souviennent peut-être des annonces spectaculaires de la société Celera Genomics en 2000. Le bouillant fondateur de Celera, Craig Venter, prétendait alors avoir obtenu une version de qualité du génome humain par la stratégie du séquençage aléatoire global. Du fait de ces annonces et de la polémique qui a suivi, il est intéressant de revenir sur quelques aspects de ce séquençage.
Celera a effectivement produit près de 15 Gb de lectures aléatoires à partir de l’ADN de plusieurs individus, soit une profondeur de 5X. Toutefois, nous ignorons si Craig Venter et ses collaborateurs ont tenté d’assembler ces seules séquences, et, si oui, quel a été le résultat de cet assemblage à 5X… On peut penser qu’une telle tentative était vouée à l’échec : certains chercheurs estiment qu’il faudrait une profondeur bien supérieure à 10X pour atteindre une couverture satisfaisante du génome humain par une stratégie de séquençage aléatoire global.
Quoi qu’il en soit, l’équipe de Celera ne s’en est pas tenue à ses seules données. Elle y a ajouté des données de séquence « empruntées » au consortium public, le plus légalement du monde, puisque ces données, contrairement aux leurs, sont librement accessibles aux chercheurs du monde entier. Pour effectuer cet emprunt, Venter et ses collègues ont cassé informatiquement, de deux façons différentes, les séquences assemblées par le consortium ; ils ont ainsi produit des pseudo-lectures régulièrement réparties et parfaitement chevauchantes, dont ils affirment qu’elles ne représentent « que » 2,9X – un petit appoint, selon eux, à leurs 5X de séquence. En fait, ces pseudo-lectures correspondent aux 7,5X de séquence publique qui avaient servi à assembler la séquence « décomposée » : elles retiennent l’essentiel de l’information d’assemblage. L’« emprunt » est donc bien plus conséquent que Venter veut bien le reconnaître. Mais l’utilisation des données du public ne s’est pas arrêtée là : Celera s’est également servi de la carte physique établie par le consortium international pour réaliser un assemblage « compartimenté ». Diviser la difficulté de l’assemblage en regroupant localement les lectures : une philosophie assez éloignée du séquençage aléatoire global.
Quel a été le résultat de tous ces emprunts ? Étonnamment, la séquence assemblée par Celera et décrite dans un article de la revue Science, en février 2001, n’était pas spectaculairement meilleure que celle du consortium public, comme on aurait pu l’attendre. Elle ne couvrait que 90 % du génome humain, le reste étant sous la forme de millions de lectures d’environ 600 bases, inutilisables car non assemblées. Et surtout, il demeurait près de 170 000 trous…
Toutefois, ces querelles de chiffres ont pesé peu, à l’époque, face aux effets d’annonce qui ont fait grimper l’action de Celera.
Il faut reconnaître que cette initiative privée a servi d’aiguillon au projet public : c’est en partie à Craig Venter que l’on doit de disposer, depuis avril 2003, d’une version de référence de la séquence du génome humain, gratuite et accessible sans restriction.
Stratégie « clone par clone »
Cette stratégie « clone par clone » (dite encore du « shotgun hiérarchique ») est celle qui a été adoptée par le consortium international pour le séquençage du génome humain (HGP : Human Genome Project). Il s’agit d’une démarche en deux temps : établissement d’une carte physique, ordonnant des clones de grande taille dans le génome humain, puis séquençage (de type « shotgun ») de ces clones. La carte peut aussi être construite en même temps que le séquençage progresse. Une aide essentielle dans la construction d’une carte physique est apportée par les cartes de liaison.
Les cartes de liaison : pour se repérer dans le génome
Ces cartes permettent de disposer de marqueurs, c’est-à-dire de points de repère, ordonnés le long des chromosomes par la mesure de leur liaison deux à deux. La nature de cette liaison dépend de la nature de la carte. On utilise deux types d’approches pour construire une carte de liaison.
Une première approche est l’utilisation de marqueurs génétiques polymorphes, qui sont ordonnés grâce à l’étude des fréquences de recombinaison génétique (mesure de leur « liaison génétique »). Les cartes obtenues de cette manière sont nommées « cartes génétiques ». La première carte génétique de l’ensemble du génome humain remonte à 1987 ; elle reposait sur des marqueurs de type RFLP, obtenus grâce aux enzymes de restriction. Les marqueurs privilégiés depuis le début des années 90 sont les microsatellites. En 1996, le laboratoire Généthon a publié une carte génétique de référence du génome humain, ordonnant 5264 microsatellites, qui est encore très utile aujourd’hui. Elle a notamment permis de cartographier de nombreux gènes associés à des maladies génétiques.
Dans le second type de cartes de liaison, on utilise des marqueurs moléculaires non nécessairement polymorphes. Il s’agit de séquences d’ADN présentes de manière unique dans le génome ( STS : Sequence Tagged Sites), ce qui inclut aussi les marqueurs génétiques. On ordonne ces marqueurs en mesurant la fréquence avec laquelle deux d’entre eux sont séparés par une cassure induite par rayons X. On parle dans ce cas de cartes obtenues par hybrides d’irradiation.
Les marqueurs moléculaires des cartes de liaison sont précieux pour valider et ancrer les cartes physiques le long des chromosomes.
La carte physique : une collection de clones
L’établissement d’une carte physique a principalement pour but de faciliter l’établissement de la séquence finale du génome. Dans la stratégie du séquençage « clone par clone », la phase de séquençage aléatoire est conduite sur chacun des grands fragments ordonnés de la carte, et non sur l’ensemble du génome. Cela permet de réduire la difficulté d’assemblage à des fragments de 300 milliers de paires de bases au maximum, au lieu des 3 milliards du génome entier. Cette stratégie permet aussi de focaliser le travail de finition : on peut repartir à volonté du fragment sur lequel on travaille pour parfaire le séquençage, boucher les trous, etc. Il est en outre plus facile de répartir le travail entre plusieurs collaborateurs avec un minimum de coordination, de vérifier la validité de la séquence assemblée, et d’éviter en partie les problèmes posés par le polymorphisme (dans un séquençage aléatoire global, même en partant d’un seul individu, on assemble en effet des séquences qui proviennent de deux chromosomes).
Pour construire une carte physique, on casse le génome (humain dans notre cas) en fragments de grande taille, afin de couvrir l’ensemble du génome avec relativement peu de fragments. La carte construite par le consortium international a permis ainsi de définir un « chemin de recouvrement minimal » de 26 614 fragments, pour un total de 2 841 366 484 paires de bases.
Les fragments nécessaires à la réalisation d’une carte physique mesurent en moyenne plus de 100 000 paires de bases (100 kilobases). Le premier problème qui s’est posé à la communauté scientifique a été de trouver des vecteurs supportant des inserts d’une telle taille.
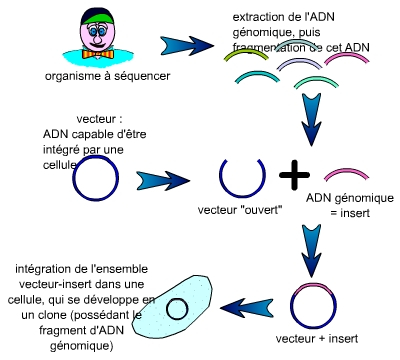
L’ADN génomique est fragmenté. Chaque fragment est inséré dans un vecteur, puis l’ensemble vecteur-insert est intégré dans une cellule (bactérie, levure…) qui, après étalement, se multiplie pour former une population de cellules toutes identiques (un clone), visible sous la forme d’une colonie.
On constitue alors une collection de clones cellulaires ayant intégré un couple insert-vecteur. Cette collection est nommée « banque d’ADN génomique », puisque ces clones constituent une représentation, que l’on espère la plus complète possible, de l’ADN génomique de l’organisme à séquencer.
Les vecteurs les plus utilisés en biologie moléculaire, plasmides et cosmides, ne permettent pas de cloner des fragments de plus de 45 kilobases (45 000 paires de bases). Ils sont donc inadaptés au clonage d’inserts suffisamment grands pour réaliser une carte physique. Dans un premier temps, les chercheurs ont utilisé les chromosomes artificiels de levure ( YAC : Yeast Artificial Chromosome). Ces vecteurs permettaient l’insertion de fragments allant jusqu’à 1 000 kilobases. Mais ils ont été abandonnés : en effet, des échanges de fragments d’ADN avaient lieu… Certains clones « chimériques » ne correspondaient ainsi plus aux inserts clonés.
Les vecteurs qui ont été utilisés pour établir la carte physique du génome humain sont principalement les chromosomes artificiels bactériens ( BAC : Bacterial Articifial Chromosome). Bien que de plus faible capacité que les YAC (seulement 300 kilobases au maximum, 150 kilobases en moyenne), ils n’en présentent pas les graves défauts. Des vecteurs dérivés du phage P1 (les PAC), de même capacité, ont aussi été utilisés.
Ordonner les clones
La première étape a donc consisté à obtenir des clones de grande taille, de l’ordre de 100 à 200 kilobases. Il a fallu ensuite ordonner ces clones, c’est-à-dire les positionner les uns par rapport aux autres, et le long des chromosomes humains.
Le positionnement des clones les uns par rapport aux autres fait appel à différentes techniques, dont le point commun est de rechercher les parties communes entre différents clones. Parmi ces techniques, on peut citer :
-
l’utilisation des profils de restriction : on digère les clones grâce à des enzymes de restriction, puis on recherche si différents clones présentent des fragments de même taille. Il y a alors de bonnes chances que ces clones possèdent une région génomique en commun, que les enzymes de restriction coupent aux mêmes endroits.
-
l’hybridation des clones entre eux : si deux clones peuvent s’hybrider, c’est qu’ils possèdent des séquences communes.
-
l’utilisation des STS (Sequence Tagged Sites) : si deux clones peuvent s’hybrider avec l’une de ces séquences uniques du génome humain, c’est qu’ils possèdent une région commune.
L’on peut alors positionner le long des chromosomes les groupes de grands clones chevauchants ainsi constitués, en particulier grâce aux cartes de liaison.
Une fois tous les grands clones ordonnés et positionnés le long du génome humain, on dispose d’une carte physique du génome. On peut alors sélectionner un ensemble minimal de grands clones chevauchants (un chemin de recouvrement) en vue de les séquencer. Concrètement, le choix des grands clones à séquencer peut être réalisé en même temps que le séquençage progresse, ce qui permet, dans une certaine mesure, de minimiser les régions de chevauchement entre les grands clones.
Une stratégie « shotgun » pour le séquençage final
Pour séquencer les clones de grande taille, une stratégie de type « shotgun » (séquençage aléatoire) est utilisée : chaque clone de grande taille est découpé en un grand nombre de fragments de petite taille (environ 2 kilobases – 2 000 paires de bases), dont les extrémités sont séquencées individuellement… Ces séquences sont ensuite assemblées, afin de fournir une séquence aussi complète que possible du clone de grande taille. L’assemblage des séquences des clones de grande taille (aux trous près, qui peuvent demeurer entre ces clones) donne la séquence du génome humain.
Avancement du Projet Génome Humain
La carte physique a été achevée par le consortium international début 2000. Elle couvrait alors 97 % du génome humain. Le séquençage à 7,5X à partir de cette carte avait permis d’assembler une ébauche de la séquence qui couvrait, quant à elle, 87 % du génome, dont 28 % étaient déjà sous forme de séquence « finie ».
Grâce au passage de 7,5X à 10 X et à une finition ciblée sur les zones de faible qualité et les trous, le consortium est passé de cette ébauche à une séquence complète du génome humain, célébrée en avril 2003. Cette séquence couvre 99 % du génome humain, avec une précision de 99,99 %.
Documents à télécharger
Animation Flash (fichier swf)
Apport des génomes séquencés
À quoi bon dépenser beaucoup d’argent public ou de dons pour séquencer des génomes ?
C’est une question que l’on peut – et que l’on doit – se poser. La réponse n’est pas simple, car les séquençages complets de génomes peuvent avoir des utilités très diverses, aussi bien pour la médecine, la recherche appliquée, que pour la recherche fondamentale (sans laquelle, rappelons-le, il ne peut exister de recherche appliquée).
Nous proposons ici, en guise de conclusion de ce document sur le séquençage des génomes, quelques exemples succincts d’apports possibles des séquençages génomiques.
Connaissance des gènes
La détermination de la séquence complète d’un génome n’est que la première étape de son étude. Il est en effet nécessaire de déterminer ensuite où, exactement, se situent les gènes et leurs régions régulatrices. Cette « annotation » est toujours en cours pour le génome humain, même si de très nombreux gènes ont déjà été repérés. On peut espérer dresser dans un avenir proche un inventaire relativement exhaustif et précis des gènes humains. Le séquençage complet du génome constitue une étape nécessaire pour arriver à ce résultat. L’étude des seuls ARN messagers s’est en effet avérée rapidement insuffisante. De plus, l’obtention d’une séquence complète du génome humain permet d’éviter que les chercheurs du monde entier se lancent dans des recherches de gènes de manière redondante, et donc moins efficace et plus onéreuse.
Cette connaissance exhaustive des gènes humains est un effort de recherche fondamental. De manière directe, ces résultats n’ont pas d’application. Toutefois, cette connaissance des gènes permet, ensuite, d’aboutir à de nombreuses applications pratiques.
Par ailleurs la connaissance des génomes de plusieurs organismes permet, d’une part, de faciliter l’identification des gènes via des comparaisons entre séquences génomiques, et d’autre part, de comparer les gènes eux-mêmes. Ces recherches, qui peuvent être menées sur des gènes présents dans des organismes phylogénétiquement très distants (de nombreux programmes de séquençage étant en cours), permettent de mieux cerner la fonction et l’importance de ces gènes, ainsi que leur histoire évolutive. Ces futures découvertes devraient être profitables à toutes les branches des sciences de la vie, que cela soit le développement embryonnaire, l’immunologie, les neurosciences, etc.
Recherche liée aux maladies génétiques
De nombreuses maladies humaines sont dues à l’expression (ou au défaut d’expression) d’allèles d’un unique gène (on parle en général d’allèles « mutés », même si cette terminologie est discutable) : ce sont les « maladies génétiques ». Pour espérer soigner et surtout diagnostiquer ces maladies, il est important de savoir quel est le gène impliqué (il arrive que ce ne soit pas toujours le même gène d’un malade à un autre), et quel est l’allèle (ou les allèles) responsable(s).
Sans séquence complète du génome, la tâche est souvent très difficile : à partir d’études menées sur des familles atteintes de ces maladies, les chercheurs remontent jusqu’à une « région » chromosomique portant la mutation en question. Il faut ensuite chercher « à l’aveugle » les centaines de gènes de cette région, pour essayer de trouver le gène impliqué. Avec la séquence complète annotée, dès lors qu’une région chromosomique est impliquée, on dispose immédiatement de la liste des gènes présents dans cette région. En se basant sur les propriétés connues (ou supposées, par analogie) de ces gènes, on peut alors très rapidement orienter les études vers les quelques gènes « candidats » qui ont le plus de chance d’être impliqués dans la pathologie étudiée (il arrive toutefois que le gène affecté soit un gène qu’aucun indice ne pouvait faire suspecter, et que seule l’étude génétique pouvait donc impliquer, ce qui fait toute la puissance de cette approche du « clonage positionnel »). L’ensemble de l’analyse est ainsi plus rapide, moins cher à réaliser.
Ceci devrait donc se traduire dans un avenir proche par l’isolement de très nombreux gènes responsables de maladies génétiques. Ces découvertes pourront alors être le premier pas vers la mise au point de meilleurs traitements de ces maladies, voire de propositions de thérapies définitives. C’est ainsi qu’un traitement prometteur de l’Ataxie de Friedreich, directement issu de la connaissance du gène et de sa fonction, a été développé en 1999 par une équipe française à l’Hôpital Necker.
Diagnostics ADN
Connaître le génome humain dans son intégralité permet donc d’envisager la connaissance des allèles des gènes responsables de maladies génétiques. Ceci pourra faciliter la mise au point de test diagnostics à partir de l’ADN.
Pour les maladies les plus graves, le diagnostic génétique peut être pratiqué avant la naissance dans les familles à risque. De manière générale, la mise au point de diagnostics génétiques permet d’affiner l’identification précise de la maladie atteignant le patient ; ceci ne peut que conduire à une meilleure prise en charge de cette maladie.
Recherches de susceptibilités
Connaissant le génome humain, et après étude des positions variables d’une personne à une autre, il sera plus facile d’identifier les facteurs génétiques de susceptibilité à de nombreuses maladies.
Par exemple, des maladies comme le diabète ou l’artériosclérose, ont une composante génétique correspondant à une multitude de facteurs. Chaque facteur ne contribue à la maladie finale que pour une faible part. De plus, la maladie est en fait causée par l’interaction complexe entre ces facteurs et les conditions de l’environnement. Tout ceci fait qu’il est encore extrêmement difficile de dire à l’heure actuelle ce qui cause ces maladies…
Arriver à identifier les facteurs génétiques impliqués, et démêler cet écheveau devrait permettre de mieux comprendre la genèse de ces maladies, et par là même d’aboutir à de nouveaux traitements, à des mesures de prévention plus efficaces.
Puces à ADN et transcriptome
La connaissance de génomes complets, annotés, permet la réalisation de puces à ADN, et donc l’étude du transcriptome, au-delà de celle du génome. Les puces à ADN sont des outils permettant de mesurer et visualiser rapidement les différences d’expression entre les gènes, à l’échelle d’un génome complet. Il s’agit donc de l’étude directe et qualitative du transcriptome, c’est-à-dire de l’ensemble du matériel génétique exprimé dans une cellule donnée.
Les puces à ADN (qui n’ont strictement rien à voir avec les ordinateurs et les puces électroniques !) sont des lames recouvertes de sondes correspondant aux gènes appartenant au génome d’un organisme. Chaque sonde, donc chaque gène, est placé à un endroit précis et identifié sur la plaque. La puce permet ensuite de comparer l’expression des gènes entre deux souches de cellules (une souche sert de témoin, l’autre correspondant à l’étude menée) : pour cela, les ARN de ces cellules sont extraits, rétro-transcrits en ADN, et marqués à l’aide d’un fluorochrome (vert pour une souche, rouge pour l’autre). L’ensemble est alors incubé avec la puce à ADN : les ADNc correspondants aux ARN des cellules s’hybrident avec les sondes portées par la puce. La puce est ensuite lue, gène par gène, à l’aide d’un laser. On « lit » ainsi trois types de gènes : (1) des gènes exprimés de manière plus importante dans la première souche, dont les sondes ont fixé un plus grand nombre d’ADNc issus de la première souche, et qui fluorescent donc essentiellement en vert ; (2) des gènes exprimés de manière plus importante dans la deuxième souche, qui apparaissent plutôt en rouge ; (3) des gènes exprimés à des niveaux comparables.
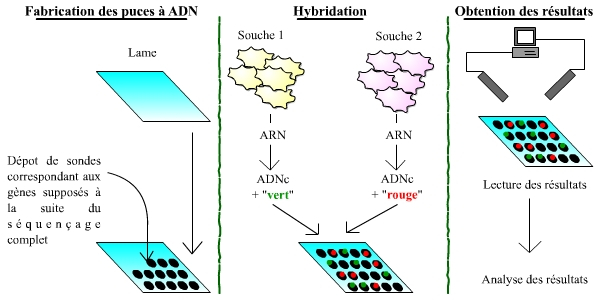
Les trois grandes étapes du processus sont schématisées : la fabrication des puces, l’hybridation, puis l’obtention des résultats.
Attention : ce schéma est extrêmement simplifié par rapport à la réalité !
De nombreuses études utilisant ces puces à ADN sont d’ores et déjà menées chez la levure, dont le génome est maintenant connu depuis plusieurs années. De telles études sont appelées à se développer chez l’Homme, ainsi que pour toutes les espèces modèles dont le génome est séquencé intégralement.
Quelques informations, illustrations, etc. sont disponibles sur le site de la plate-forme transcriptome du service de génomique du département de biologie de l’École normale supérieure. Ce service commun réalise et exploite des puces à ADN pour le compte des laboratoires de biologie de l’École normale supérieure ou d’autres laboratoires du « pôle Montagne Sainte-Geneviève », à Paris essentiellement (Institut Curie, École supérieure de physique et chimie industrielle).
Pour d'autres informations sur le séquençage du génome humain, consultez le site de l'université d'Angers.
Liens
-
Le Généthon. Des informations et des liens sur les banques d’ADN, les gènes, la recherche en thérapie génique.
-
Le Génoscope. Centre national français de séquençage.
Glossaire
-
BAC :
« Bacterial Artificial Chromosome », chromosome artificiel de bactérie. Vecteur de grande capacité (de l’ordre de 300 kilobases), possédant les séquences permettant sa réplication et son maintien dans une bactérie E. coli. -
Cosmide :
Molécule d’ADN circulaire comportant des séquences virales (et qui peut ainsi être véhiculé dans des enveloppes virales). Il permet de cloner des inserts jusqu’à 45 kilobases (45 000 paires de bases). -
Enzyme de restriction :
Enzyme capable de digérer l’ADN (endonucléase) en un endroit présentant une séquence précise, spécifique de l’enzyme. La séquence d’ADN reconnue par l’enzyme de restriction est nommée « site de restriction ». Ces sites de restriction peuvent comporter de 2 à une vingtaine de paires de bases. Exemple : l’enzyme EcoR I reconnaît et hydrolyse l’ADN au niveau des séquences GAATTC. -
Microsatellite :
Répétitions de nombreuses fois d’une courte séquence de 2 à 6 paires de bases. Ces séquences d’ADN, présentes dans l’ensemble du génome humain, sont hautement polymorphiques. -
Plasmide :
ADN circulaire de quelques dizaines de kilobases en général. On en trouve essentiellement dans les bactéries. Les vecteurs utilisés dans le clonage de gènes sont, dans leur grande majorité, des plasmides (pBR 322, pBluescript, etc.). Ils permettent le clonage d’inserts de 20 à 30 kilobases au maximum. -
Polymorphe :
Un gène existe sous la forme de nombreux allèles. Il est dit polymorphe dès que deux allèles distincts, au minimum, sont présents dans une population avec une fréquence d’au moins 1 % (chacun). Par extension, si un fragment d’ADN quelconque (séquence répétée, microsatellite, etc.) existe sous différentes formes (= différentes séquences) dans une population avec une fréquence d’au moins 1 %, il est dit polymorphe. -
RFLP :
« Restriction Fragment Length Polymorphism ». Il s’agit d’une technique consistant à digérer l’ADN génomique par des enzymes de restriction. L’existence de différences de séquences entre les individus conduit à des différences dans les sites de digestion entre ces individus. Après séparation des fragments obtenus par électrophorèse, on observe donc des fragments de tailles diverses selon les individus. Par comparaison, on dispose ainsi de marqueurs répartis dans tout le génome. -
YAC :
« Yeast Artificial Chromosome », chromosome artificiel de levure, obtenue en associant des séquences centromériques et télomériques issues de chromosomes de levures. Ces vecteurs acceptent des inserts de très grande taille (1 000 kilobases). Ils ont comme grave défaut d’être sujets à des réarrangements, une fois intégrés dans les cellules de levure.



