Cet article présente une méthode de thérapie génique appliquée en 2005 pour traiter des enfants-bulle en remplaçant le gène impliqué dans la maladie par un gène fonctionnel. Cette méthode utilise des nucléases à doigt de zinc, capables de d'induire une coupure double-brin spécifiquement au niveau du gène d'intérêt. La copie fonctionnelle du gène s'insère ensuite par recombinaison homologue.
Introduction
Pour soigner les maladies génétiques, de grands espoirs ont été placés au cours de la décennie précédente dans la thérapie génique, c’est-à-dire dans l’apport direct de gènes non mutés aux cellules qui n’en possèdent que des exemplaires mutés.
Mais à ce jour, il n’y a que pour la maladie des enfants-bulle que cette technique s’est montrée efficace, du moins jusqu’à une époque récente où des problèmes graves sont apparus, qui ont conduit à interrompre son utilisation. Cependant, une voie d’action tout à fait nouvelle vient de commencer à être expérimentée, qui dégage à nouveau l’horizon de la thérapie génique.
La maladie des enfants-bulle et les voies thérapeutiques précédentes
La maladie des enfants-bulle est une maladie très rare : cinq cas sur près de huit cent mille naissances chaque année en France. Elle se manifeste par un déficit immunitaire sévère, qui condamne les jeunes malades à vivre à l’abri des germes dans une atmosphère stérile, d’abord à l’intérieur d’une bulle de plastique, puis dans un local spécialisé. Un scaphandre doit être employé pour les sorties.
Dans sa variante la plus connue, ce déficit est dû à la présence, sur le chromosome X, d’une version mutée d’un gène codant une protéine participant à la constitution du récepteur de l’interleukine 2 (la sous-unité gamma-c) sur la membrane de certains globules blancs. L’absence du récepteur complet rend impossible l’orchestration, par l’interleukine 2, des globules blancs : lymphocytes T (LT), lymphocytes B (LB) et lymphocytes natural killer (NK).
Les garçons, qui ne disposent que d’un seul chromosome X, deviennent ainsi sujets à toutes les infections. La maladie des enfants-bulle est donc en fait, dans sa variante la plus connue, la maladie des garçons-bulle ou « déficit immunitaire combiné sévère lié au chromosome X », notée DISC-X (Figure 1).
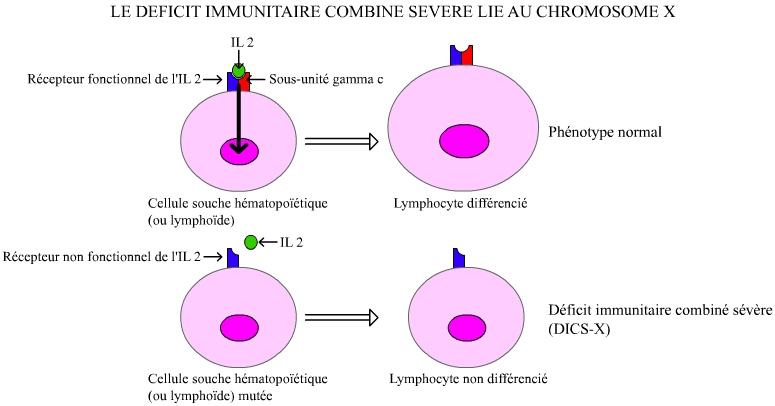
La sous-unité gamma-c intervient dans la constitution de différents récepteurs aux interleukines (IL2, 4, 7, 9 et 15). La mutation du gène codant cette sous-unité empêche la mise en place de ces récepteurs. L’absence de récepteur fonctionnel pour l’IL2 a pour conséquence une absence de différenciation des cellules souches lymphoïdes en lymphocytes, la transduction du signal ne pouvant s’effectuer.
Les seules perspectives de soin pour ces petits garçons sont soit une greffe de moelle osseuse, tissu qui est à la source des globules blancs, soit une thérapie génique qui apporte le gène non muté. Mais les donneurs de moelle compatible sont rares et la thérapie génique est interrompue actuellement par des problèmes majeurs.
Pourtant, en 1999, les premiers essais de thérapie génique, conduits par l’équipe d’Alain Fisher à l’hôpital Necker, débutent par un spectaculaire succès, qui est aussi le premier jamais obtenu par thérapie génique : dix enfants sont traités, et guérissent. Les cellules souches de leurs globules blancs avaient été prélevées dans leur moelle osseuse et traitées in vitro par un rétrovirus désactivé qui ajoutait la version non mutée du gène à leur génome porteur du gène muté. Ainsi complétées, ces cellules souches avaient alors été réinjectées dans la circulation sanguine des petits malades.
Mais voici qu’en octobre 2002, deux d’entre eux, qui avaient été traités avant l’âge de trois mois, deviennent leucémiques. L’un guérira, mais l’autre finira par en mourir. Les essais thérapeutiques sont aussitôt suspendus. Dans les deux cas, le gène correcteur s’est inséré au voisinage du même gène prédisposant au cancer, LMO-2.
Les protocoles sont alors révisés : début du traitement repoussé après l’âge de six mois, recours à des doses plus faibles de cellules modifiées. Et les essais thérapeutiques sont de nouveau autorisés en mai 2004.
Hélas, un troisième cas de leucémie se déclare en janvier 2005 chez un enfant traité à l’âge de neuf mois. Les essais thérapeutiques sont une seconde fois suspendus. Le gène médicament s’est à nouveau inséré auprès d’un oncogène (qui n’est pas LMO-2). Les essais ne pourront reprendre à l’hôpital Necker que lorsque le virus vecteur du gène salvateur aura pu être modifié.
Parallèlement, en Grande-Bretagne, les sept enfants traités selon un protocole pratiquement similaire au premier protocole français sont tous guéris. Cependant, le temps écoulé depuis le traitement n’a pas encore atteint la durée à partir de laquelle les leucémies françaises sont survenues. Enfin, des essais vont être conduits pour la première fois aux États-unis. Ils s’adresseront aux enfants sur lesquels une greffe de moelle osseuse a échoué ou n’a pu être tentée.
C’est dans ce contexte qu’une nouvelle voie de thérapie génique vient de commencer à être expérimentée.
La maladie des enfants-bulle et la nouvelle voie de thérapie génique
Cette nouvelle voie thérapeutique est due à Fyodor D. Urnov et ses collaborateurs, de Sangamo BioSciences en Californie et de l’université Southwestern au Texas. Sa nouveauté fondamentale est qu’elle opère par remplacement et non par addition, c’est-à-dire que la portion mutée du gène a été précisément remplacée par la version non mutée, sans toucher au reste du génome. Dans le processus antérieur, la version non mutée du gène était ajoutée à un génome qui demeurait porteur de la version mutée d’origine.
Pour effectuer cette substitution, les chercheurs ont opéré sur les lymphocytes T prélevés chez les malades et ont réussi in vitro l’admirable enchaînement suivant :
-
Détection du site précis de la mutation par des protéines à doigts de zinc.
Les doigts de zinc sont des structures de reconnaissance de l’ADN (Figure 2). En fonction de la nature des acides aminés qui les composent (hors les cystéines et histidines liant l’atome de zinc), une séquence spécifique d’ADN de trois nucléotides sera reconnue.
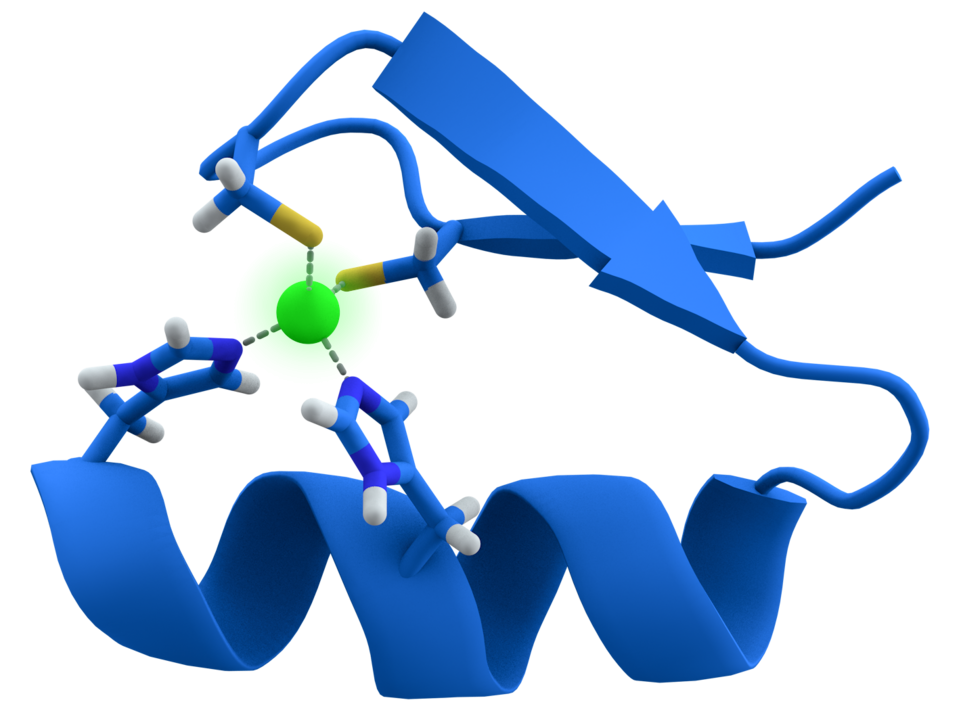
Un atome de zinc central stabilise le repliement de la protéine en établissant quatre liaisons avec deux résidus cystéine (en haut) et deux résidus histidine (en bas). On trouve également des structures en doigt de zinc faisant intervenir quatre résidus cystéine.
En utilisant deux protéines comportant chacune trois doigts de zinc pouvant être individuellement choisis, la reconnaissance s’effectue sur une séquence spécifique de dix-huit nucléotides (Figure 3). Un rapide calcul de probabilité établit qu’une séquence aléatoire de dix-huit nucléotides n’a pratiquement aucune chance d’exister à plus d’un exemplaire dans un génome de six milliards de nucléotides correspondant au génome humain. En conséquence, par cette méthode on peut cibler de manière spécifique et unique une séquence d’intérêt connue (par exemple un gène muté…).
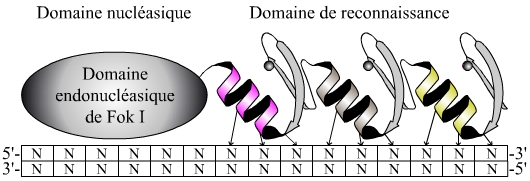
Chaque doigt de zinc reconnaît trois nucléotides spécifiques. Une protéine comportant trois doigts de zinc en tandem permet donc de reconnaître une séquence spécifique de neuf nucléotides. À ce domaine de reconnaissance est couplé le domaine endonucléasique de l’enzyme Fok I.
-
Coupure de l’ADN ciblé par une enzyme (une endonucléase) préalablement liée à deux protéines en doigts de zinc.
L’endonucléase utilisée (domaine endonucléasique de l’enzyme Fok I) est non spécifique et agit sous forme dimérique. On utilise donc deux protéines comportant chacune trois structures en doigt de zinc reconnaissant des séquences éloignées de quelques nucléotides. La liaison des deux protéines en doigts de zinc sur leurs séquences respectives rapproche les deux endonucléases qui leur sont associées. Ce rapprochement permet leur dimérisation et par suite la coupure de la molécule d’ADN (Figure 4).

Chaque nucléase à doigts de zinc (ZFN) reconnaît une séquence de neuf nucléotides séparés par six nucléotides, un espace nécessaire pour l’action de l’endonucléase. Cette action n’est possible qu’après dimérisation de cette dernière.
-
Exploitation du mécanisme naturel de recombinaison homologue pour remplacer la portion du gène muté par la portion équivalente du gène non-muté.
La courte molécule d’ADN non mutée, susceptible de remplacer la fraction mutée à l’origine de la maladie, est ajoutée aux deux molécules protéiques précédentes (Figure 5). Si le phénomène de recombinaison homologue (largement utilisée dans la technique d’inactivation de gène, plus connue sous le nom de knock-out) ne nécessite pas une coupure préalable de l’ADN par endonucléase, son efficacité dans ces conditions est très faible. Couper l’ADN permet d’augmenter considérablement le taux de recombinaison homologue.
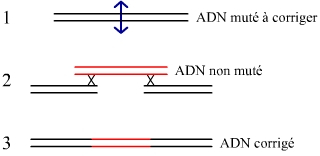
La molécule d’ADN ajoutée comporte le gène non-muté à insérer encadré de séquences flanquantes identiques aux séquences qui bordent le site de coupure par l’endonucléase. La recombinaison se produit de façon spontanée.
Dans près de 20 % des cas – un taux énorme pour une recombinaison homologue, et un taux suffisant pour soigner plus tard les malades – la portion d’ADN mutée des lymphocytes T a été remplacée par sa version non mutée. Le gène corrigé s’exprime sous forme d’ARNm, puis de la protéine gamma-c permettant la bonne mise en place du récepteur de l’interleukine 2. La correction s’effectue de façon rapide et se maintient de façon durable.
Ultérieurement, ce ne sont pas les lymphocytes T, les lymphocytes B ou les lymphocytes NK que les chercheurs corrigeront in vitro par cette méthode, mais leurs cellules souches progénitrices logées dans la moelle osseuse qui bénéficient d’une longue durée de vie. Une fois traitées, elles seront réinjectées dans la circulation des petits patients pour pouvoir coloniser leur moelle osseuse.
Conclusion
Cette nouvelle voie de thérapie génique est riche de promesses parce qu’elle pourrait, sans affecter le reste du génome, et sans risquer notamment de perturber un oncogène, corriger de façon parfaitement ciblée la version indésirable d’un gène. Il faut cependant que soit garantie l’absolue spécificité de la méthode vis-à-vis du gène à corriger, ou encore l’absence de réactions immunitaires provoquées par les protéines à doigts de zinc qui sont les artisans de ce ciblage.
Il deviendrait alors possible d’étendre cette technique à d’autres maladies, car c’est aujourd’hui toute une batterie de ces protéines à doigts de zinc que les chercheurs savent fabriquer à la demande, et qui sont à chaque fois capables de reconnaître une cible différente : qu’il s’agisse du gène muté dans le cas d’autres maladies monogéniques, comme l’anémie falciforme, ou qu’il s’agisse du gène à faire muter, comme celui codant la porte d’entrée du virus du sida à l’intérieur des lymphocytes T !
Bibliographie
-
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD et Holmes MC. « Highly efficient endogenous human gene correction using designed zinc-finger nucleases. » Nature. 435(7042) :577-579. 2005-06.