Les facteurs environnementaux et le métabolisme de la cellule elle-même produisent de 1 000 à 1 000 000 lésions de l'ADN par cellule et par jour. Cet article présente les types de dommages que peut subir l'ADN et les mécanismes que possèdent les cellules pour les corriger. Il développe en particulier le mécanisme de réparation des cassures double-brin par recombinaison homologue.
Introduction
Bien qu’isolé pour la première fois en 1869 par Friedrich Miescher, ce n’est qu’au milieu du vingtième siècle que l’importance de l’acide désoxyribonucléique (ADN) est découverte. En 1944, Oswald T. Avery, Colin MacLeod et Maclyn McCarthy montrent que l’ADN d’une souche bactérienne pathogène est le seul élément nécessaire et suffisant pour permettre la transformation d’une souche non pathogène en souche pathogène. Alfred Hershey et Martha Chase observent plus tard que l’ADN d’un bactériophage, mais pas ses protéines, pénètre dans les bactéries infectées et est retrouvé dans les phages créés après l’infection. Ces travaux montrent que l’ADN est le support de l’information génétique. La compréhension de la structure de l’ADN par Watson et Crick en 1953, confirmée par les images de diffraction aux rayons X de l’ADN obtenues par Rosalind Franklin et Maurice Wilkins, montre que l’ADN est formé de deux brins complémentaires associés de manière antiparallèle en une double hélice (Franklin & Gosling, 1953 ; Watson & Crick, 1953a ; Watson & Crick, 1953b ; Wilkins et al, 1953). Cette découverte a valu un prix Nobel à Watson, Crick et Wilkins en 1962. Cette structure si particulière, qui utilise la complémentarité deux à deux des bases azotées, est essentielle pour la bonne conservation et la transmission de l’information génétique.
Le déchiffrage du code génétique, par lequel des codons de trois nucléotides codent pour un acide aminé, a valu en 1968 le prix Nobel à ses découvreurs. Cette complémentarité entre séquence d’ADN et séquence protéique révèle l’importance de conserver strictement et transmettre à l’identique la séquence nucléotidique, afin de toujours traduire la même protéine. Cette idée originelle d’ « un gène, une protéine » ne reflète pourtant pas la complexité de l’information génétique. Toutes les séquences d’ADN ne codent pas pour une protéine, certaines peuvent coder pour plusieurs protéines alternatives, ou avoir d’autres fonctions, comme dans la synthèse de l’ARN non codant ou dans la structuration des chromosomes.
Pourtant, cet ADN, si important pour la vie cellulaire, est fragile. Il peut être soumis à de nombreux stress naturels au cours de la vie de la cellule, qu’ils soient endogènes ou exogènes. Ce document fera une présentation des dommages que peut subir l’ADN et des mécanismes que les cellules possèdent pour les corriger. Ce document présentera en particulier le mécanisme de réparation des cassures double brin par recombinaison homologue.
Diversité des réponses aux dommages
Nature et origine des dommages à l’ADN
Le métabolisme cellulaire normal produit entre autre des espèces réactives de l’oxygène, sources de nombreux dommages. D’autre part, l’environnement soumet en permanence un organisme à différentes agressions, tels le rayonnement ultraviolet (UV), les radiations ionisantes ou des agents génotoxiques. Parfois, des molécules toxiques pour l’ADN sont utilisées comme outil thérapeutique, notamment en chimiothérapie. Les lésions ainsi générées sont de natures très diverses : bases altérées ou perdues, liens intra – ou inter-brins, dimères de thymines, cassures simple ou double brin (Fig. 1).

Il est intéressant de noter que le métabolisme de l’ADN lui-même est source de dommages. Ainsi, les polymérases, qui réalisent la copie à l’identique de l’ADN lors de la réplication, commettent parfois des erreurs qui rompent la complémentarité entre les deux brins.
Le changement de type sexuel chez les levures peut passer par une coupure ciblée et contrôlée de l’ADN. Par exemple, dans les souches naturelles de S. cerevisiae, l’endonucléase HO produit une coupure double brin au niveau du site d’expression du type sexuel pour entraîner le changement de type sexuel. Chez Schizosaccharomyces pombe, une coupure monobrin localisée du squelette sucre-phosphate de l’ADN sert de marque pour initier le changement de type sexuel.
Ainsi, de nombreux dommages peuvent survenir au niveau de l’ADN. S’ils sont laissés tels quels, les conséquences peuvent être graves pour la cellule et ses descendantes, en fonction de la nature de la lésion. Les lésions nucléotidiques peuvent résulter en des mutations ponctuelles transmises à la descendance, ce qui est à l’origine de nombreuses maladies génétiques.
Par exemple, de nombreuses mutations ponctuelles dans le gène de la dyskérine ont été identifiées comme responsables de la dyskératose congénitale, une maladie liée à des problèmes télomériques. Une cassure double brin non réparée peut, quant à elle, résulter en la perte de la portion du chromosome ne comportant pas de centromère lors de la division cellulaire, perte délétère pour la cellule. Elle peut ainsi entraîner une aneuploïdie (nombre anormal de chromosomes dans une cellule) ou des translocations entre régions de chromosomes, ce qui peut rapidement affecter la viabilité cellulaire en supprimant ou modifiant une partie de l’information génétique disponible.
Des réparations adaptées à chaque type de dommage
De nombreux mécanismes de réponse ont été développés afin de réparer spécifiquement chacun de ces types de lésions, en fonction de la phase du cycle cellulaire (voir le document « La régulation du cycle cellulaire »). De plus, lorsque la lésion n’est pas réparée avant l’arrivée de la fourche de réplication, une voie de tolérance aux dommages existe afin d’empêcher un arrêt intempestif de la réplication (Fig. 1). Ainsi, la réparation de l’ADN comprend l’ensemble des voies permettant de rétablir l’intégrité de l’information génétique.
Réponses aux dommages nucléotidiques et simple brin
La première preuve de l’existence de mécanismes de réparation de l’ADN a été découverte en 1949, en parallèle par Albert Kelner et Renato Dulbecco. Ceux-ci ont observé que des cultures de bactéries Streptomyces griseus ou de bactériophages (virus infectant des bactéries) présentaient une meilleure survie après exposition aux UV si elles étaient exposées à la lumière du jour, juste après l’exposition aux UV (Dulbecco, 1949 ; Kelner, 1949). Il fut montré par la suite que ce phénomène est lié à la résolution des dimères de pyrimidine créés par les UV dans l’ADN, avec l’intervention d’une enzyme de photoréactivation qui utilise la lumière comme source d’énergie. Ainsi, les mécanismes de réparation de l’ADN sont spécialisés par type de dommage.
Deux voies principales conservées permettent la réparation des dommages simple brin chez les Eucaryotes (Fig. 1). La voie de réparation par excision de base passe par l’élimination du seul nucléotide endommagé. Des glycosylases spécialisées reconnaissent et éliminent la base endommagée, puis le site apurique ou apyrimidique résultant est clivé par l’AP endonucléase (Apurinic/apyrimidinic endonuclease). Un ou plusieurs nouveaux nucléotides sont alors ajoutés, puis l’ADN est ligaturé et restauré dans son état initial.
La voie de réparation par excision de nucléotides reconnaît les dommages qui déforment l’ADN et élimine un fragment de 25 à 30 nucléotides du brin d’ADN contenant la lésion. Les polymérases réplicatives et certains facteurs associés sont alors nécessaires pour re-synthétiser le fragment éliminé. Les personnes présentant des mutations dans les protéines de cette voie sont atteintes de Xeroderma pigmentosum (maladie plus connue sous le nom « enfants de la lune ») : elles sont extrêmement sensibles aux UV et développent des cancers en réponse à l’exposition au soleil car leurs cellules ne réparent pas correctement les dommages à l’ADN dus aux UV solaires. Ceci montre l’importance des protéines de cette voie dans la réparation des lésions induites par les UV.
Réponse aux lésions réplicatives
Lors de la réplication, l’ensemble du génome doit être recopié fidèlement, sans aucune erreur. Bien que les polymérases réplicatives soient extrêmement fidèles, notamment grâce à un mécanisme de vérification des erreurs, elles laissent tout de même quelques erreurs. Ainsi, chez la levure Saccharomyces cerevisiae, la polymérase réplicative delta a une fréquence de mésappariements de l’ordre de 10-8, soit une erreur tous les 100 millions de bases pour un génome d’environ 12 millions de bases. De plus, les régions contenant de petites répétitions de type microsatellites sont sources de petites insertions et délétions, augmentant la fréquence d’erreurs de la polymérase delta vers 10-3.
Ces mésappariements et insertions/délétions créent des déformations de l’ADN, qui sont reconnues par un hétérodimère de protéines spécialisé. L’erreur peut alors être excisée grâce à la reconnaissance du brin originel, et la séquence ainsi éliminée est re-synthétisée grâce à l’ADN polymérase delta et la ligase I. Ainsi, la polymérase delta intervient dans la correction de ses propres erreurs. Il est intéressant de noter que cette voie de réparation des mésappariements agit de manière couplée avec la réplication, ce qui lui permet de différencier le brin naissant du brin originel.
D’autre part, si les erreurs non réparées des ADN polymérases sont invisibles pour celles-ci lors de la réplication, les dommages simple et double brin sont, quant à eux, susceptibles d’arrêter les fourches de réplication, ce qui serait délétère pour la cellule. La voie de tolérance aux dommages, décrite par la suite, permet aux fourches de réplication de dépasser ces dommages sans les réparer (Fig. 1).
Réponse aux cassures double brin
Les cassures double brin (CDB) sont des dommages à fort risque pour la cellule, puisqu’elles peuvent entraîner la perte d’un fragment de chromosome. Deux mécanismes principaux peuvent réparer ces cassures, via deux activités différentes (Fig. 1). La réparation par jonction des extrémités non-homologues (JENH) ligature directement les deux extrémités de la cassure, alors que la recombinaison homologue (RH) utilise une séquence homologue pour recopier la région perdue lors de la cassure. Cette méthode de réparation par RH sera décrite plus loin, de manière plus détaillée.
Bien que très précis pour des cassures propres, la jonction des extrémités non-homologues est source d’erreurs si les extrémités doivent être traitées. Au contraire, la recombinaison homologue est sans erreur si elle utilise la bonne séquence homologue. Ainsi, on observe que l’activité de la jonction des extrémités non-homologues est limitée à la phase G1 des cellules haploïdes chez S. cerevisiae, lorsque aucune séquence homologue n’est disponible.
D’autres voies alternatives de réparation des CDBs existent, notamment une voie de jonction des extrémités médiée par la micro-homologie, qui est cependant source d’erreurs.
Les voies de signalisation des dommages
La reconnaissance d’un dommage à l’ADN met en place, en parallèle de la réparation, une voie de signalisation qui optimise les conditions cellulaires pour la réparation du dommage (Fig. 2). Chez S. cerevisiae, elle implique l’activation de deux protéines kinases centrales, Tel1 (ATM chez les Mammifères) et Mec1 (ATR chez les Mammifères). Le point de contrôle Tel1 reconnaît de préférence les extrémités franches des cassures double brin. Par contre, le point de contrôle Mec1 est activé préférentiellement par l’ADN simple brin. Rappelons, qu’excepté au brin discontinu des fourches de réplication et aux télomères (voir l'article sur les télomères), l’ADN simple brin n’est pas une structure ordinaire dans la cellule. Il est souvent un intermédiaire pour la réparation des dommages, notamment par les voies d’excision de base et de nucléotides.
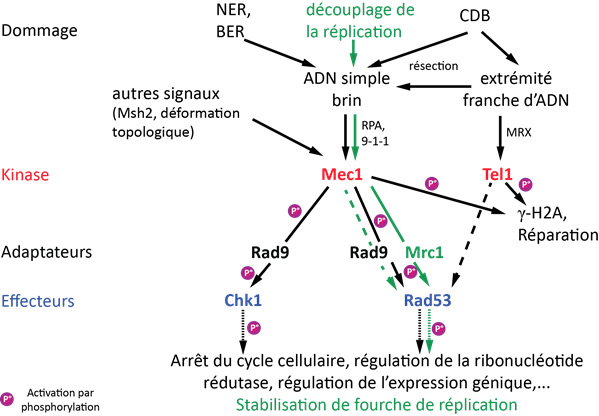
Les principaux constituants de la voie de signalisation en réponse à un dommage à l'ADN chez S. cerevisiae sont représentés, ainsi que les évènements de phosphorylation. L'apparition d'un dommage à l'ADN crée des structures qui permettent l'activation des points de contrôle cellulaires Mec1 et Tel1. Ceux-ci activent la transduction du signal par une cascade de phosphorylations. En vert : la voie de signalisation activée en cas de dommage réplicatif. Les tirets indiquent une voie secondaire.
D'après : Alcasabas et al, 2001; Harrison & Haber, 2006.
L’activation de Tel1 et Mec1 permet la phosphorylation de l’histone H2A sur plusieurs milliers de bases autour de la cassure, ce qui favorise la réparation. L’activation de Mec1 entraîne alors la transduction du signal via une cascade de phosphorylations ce qui conduit à une grande variété de réponses. L’arrêt du cycle cellulaire en G2/M est essentiel pour permettre la réparation des cassures double brin par recombinaison homologue, un processus qui nécessite plusieurs fois la durée normale d’une division cellulaire. De plus, le pool de nucléotides dans la cellule est modifié et l’expression de nombreux gènes est changée. Cette signalisation permet aussi la stabilisation des fourches de réplication en phase S. Ces nombreuses réponses mettent en place les conditions cellulaires favorables à la réparation, et Mec1 est l’acteur central de cette cascade de signalisation.
Un mécanisme similaire est mis en place par les homologues de ces protéines chez les Mammifères et implique notamment p53, un suppresseur de tumeur central dans la signalisation des dommages à l’ADN.
Une fois le dommage réparé, un processus de récupération se met en place. La réparation de la lésion entraîne l’arrêt de sa reconnaissance comme un dommage et, accompagnée d’une désactivation de la cascade de signalisation par déphosphorylation de ses composants, permet à la cellule de retrouver un cycle cellulaire normal. Tout au long de la signalisation et de la récupération, le signal est régulé finement grâce à la dégradation de ses composants par le système ubiquitine-protéasome (voir l’article « La voie de dégradation ubiquitine-dépendante »). Parfois, quand le dommage ne peut être réparé, un processus similaire d’adaptation permet à la cellule de redémarrer son cycle cellulaire pour quelques divisions, avant que les conséquences directes du dommage non-résolu n’entraînent la mort cellulaire.
Ainsi, la voie de signalisation des dommages permet de coordonner l’ensemble de la réponse cellulaire à la présence d’une lésion à l’ADN.
La recombinaison homologue
Parmi l’ensemble des lésions pouvant survenir, les cassures double brin sont des dommages très risqués pour les cellules. Un des mécanismes majeurs de réparation de ces cassures est la voie de la recombinaison homologue (Fig. 3). Cette voie, très conservée entre les différents organismes, utilise une séquence homologue à la région de la cassure pour permettre une réparation fidèle des cassures double brin.
Cette figure présente quelques molécules impliquées dans la réponse coordonnée aux cassures double brin par la voie de la recombinaison homologue.
Pour une présentation plus détaillée des mécanismes mis en jeu, voir ci-dessous.
D'après : Finn et al, 2012 ; Krogh & Symington, 2004 ; San Filippo et al, 2008 ; Symington & Gautier, 2011.
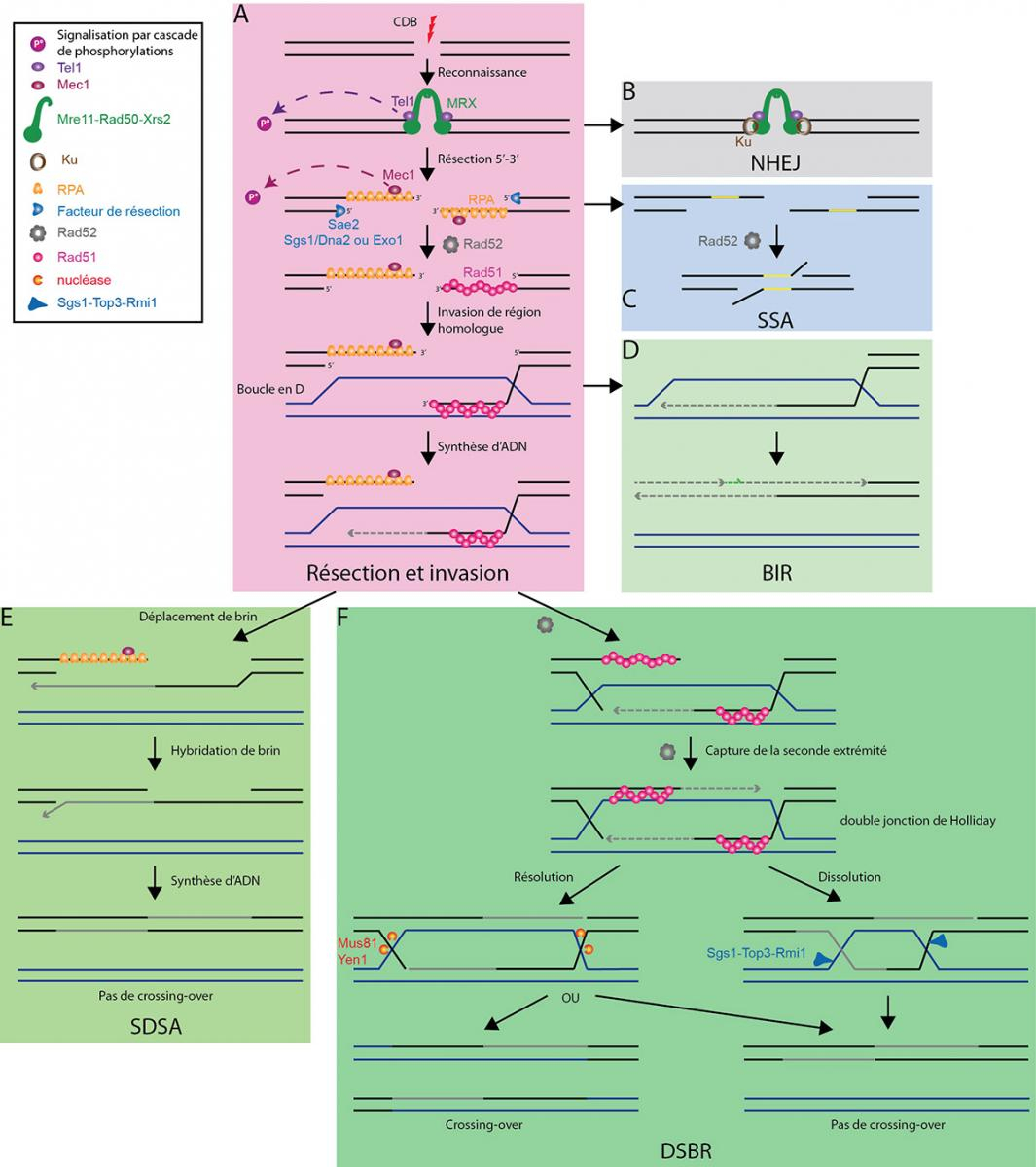
Une cassure double brin d'ADN est reconnue par le complexe MRX (A), ce qui permet l'activation de la voie de signalisation et, soit la réparation par jonction des extrémités non homologues (NHEJ) (B), soit l'initiation de la résection. Celle-ci permet alors, soit la réparation par hybridation simple brin (SSA) (C), soit la formation d'un filament Rad51-ADN simple brin. En l'absence d'une seconde extrémité, la réparation initie la réplication induite par les cassures (BIR) (D). En présence de la seconde extrémité, après synthèse d'ADN, la boucle en D formée par l'invasion d'une région homologue par le filament Rad51-ADN simple brin peut être résolue soit par déplacement de brin-hybridation de brin (SDSA) (E), soit par la voie de réparation des cassures double brin (DSBR) (F).
D'après : Finn et al, 2012 ; Krogh & Symington, 2004 ; San Filippo et al, 2008 ; Symington & Gautier, 2011.
CDB : cassure double brin
NHEJ : jonction des extrémités non homologues (Non-homologous End Joining)
SSA : hybridation de simple brin (Single Strand Annealing)
BIR : réplication induite par les cassures (Break Induced Replication)
SDSA : déplacement de brin-hybridation de brin (Synthesis-Dependant Strand Annealing)
DSBR : réparation des cassures double brin (Double-Strand Breaks Repair)
Reconnaissance du dommage et résection
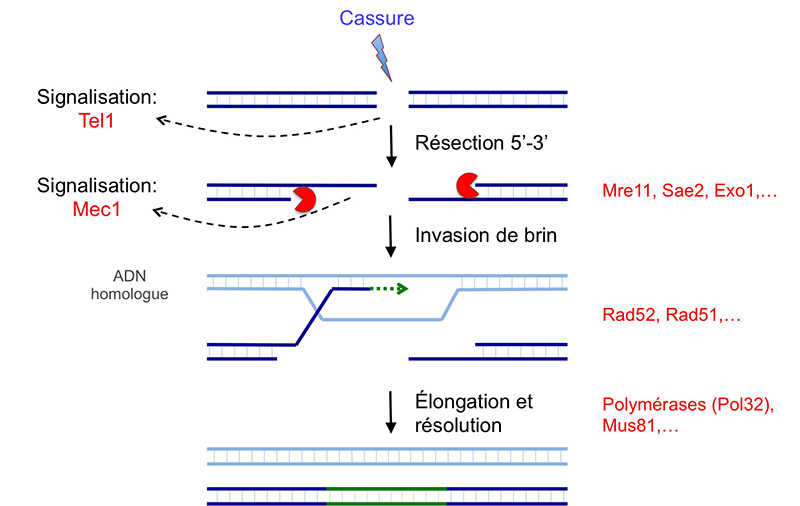
L’initiation de la réparation nécessite dans un premier temps la reconnaissance du dommage. La reconnaissance initiale d’une cassure double brin passe par le complexe MRX chez Saccharomyces cerevisiae (MRN chez l’Homme), capable de se lier à l’ADN et permet de garder attachées les deux extrémités de la cassure. Cette reconnaissance des cassures est essentielle à la fois pour l’initiation de la réparation, mais aussi pour la signalisation du dommage. Les mutants des gènes codant pour les trois protéines du complexe présentent une sensibilité exacerbée aux agents mutagènes, notamment à l’irradiation, source de cassures double brin.
Avec l’aide de MRX, une endonucléase initie la résection 5’-3’ (digestion progressive de l’un des deux brins d’une molécule d’ADN) au niveau de ces cassures, ce qui élimine environ 100 nucléotides de l’extrémité 5’ des cassures (Fig. 4A). Le choix du mécanisme de réparation des cassures double brin (jonction des extrémités non-homologues ou recombinaison homologue) est régulé à ce niveau, par le contrôle de l’initiation de la résection. L’inhibition de la résection favorise la jonction des extrémités non-homologues, alors qu’une résection étendue permet la recombinaison homologue.
Une fois initiée, la résection peut être étendue sur plusieurs kilobases par deux voies alternatives (Fig. 4A). Ces deux voies sont complémentaires et indépendantes, mutuellement exclusives (chez S. cerevisiae ; chez l’Homme il existe des interactions entre ces deux voies), et l’une peut compenser l’absence de l’autre.
Invasion d’une séquence homologue et synthèse d’ADN
Une fois formé, ce simple brin d’ADN 3’ va reconnaître une région d’homologie, qui sera utilisée pour allonger ce brin au-delà de la cassure (Fig. 3).
Recherche d’homologie et invasion
Le gène RAD51, induit en réponse aux UV et dont les mutants sont hypersensibles à l’irradiation, code pour une recombinase avec une activité de liaison à l’ADN et une activité ATPase. La protéine Rad51 couvre la longue région d’ADN simple brin formée par résection au niveau d’une cassure double brin (Fig. 4A). Une fois assemblé, le filament Rad51-ADN simple brin recherche des régions d’homologie dans le noyau. Cette recherche d’homologie est favorisée par l’augmentation de la mobilité nucléaire de la région de la cassure, ainsi que des autres chromosomes. Néanmoins, l’architecture nucléaire influence l’efficacité de la recombinaison, favorisant la recombinaison entre régions physiquement proches. Ceci avantage la recombinaison avec le chromosome homologue ou la chromatide-sœur, ce qui permet de réparer fidèlement la séquence d’ADN endommagée. La reconnaissance d’une région homologue permet la formation d’une boucle en D, par l’appariement du simple brin 3’ sortant de la cassure avec son brin complémentaire sur la région homologue, et le déplacement du second brin de la région homologue (Fig. 4A).
Polymérases et élongation
Une fois la région homologue envahie, le brin 3’ envahissant est allongé par synthèse d’ADN en utilisant le brin envahi comme matrice (Figure 4A).
Lorsqu’une des extrémités de la cassure double brin est perdue, un mécanisme de réparation particulier, appelé réplication induite par les cassures, peut se mettre en place (Figure 4D). Dans ce cas, l’extrémité de la cassure qui a envahi une région homologue sert d’initiation à un évènement de synthèse d’ADN similaire à la réplication, qui peut s’étendre sur plusieurs dizaines de kilobases. Cette copie résulte en une perte d’hétérozygotie importante, puisque la région homologue est copiée sur une grande distance. Cependant, à la différence de la réplication classique, cette voie est source de mutations. Elle implique des évènements successifs de changement de brin matrice par dissociation de la boucle en D et ré-invasion de la région homologue, avant la mise en place d’une élongation stable. Ceci permettrait de favoriser les autres voies de réparation des cassures double brin, notamment en permettant la ré-association du brin allongé avec la seconde extrémité de la cassure si celle-ci est disponible (voir ci-après).
Ainsi, l’extrémité 3’ d’une cassure double brin est allongée par synthèse d’ADN à l’aide d’une région homologue utilisée comme matrice, ce qui peut mettre en place une forme particulière de réplication, jusqu’à l’extrémité du chromosome, si d’autres mécanismes de résolution, plus fidèles, ne sont pas mis en place.
Résolution des intermédiaires formés
Une fois la synthèse d’ADN étendue au-delà de la région de la cassure, différents mécanismes sont possibles pour restaurer l’intégrité des deux chromosomes impliqués dans la réparation, selon le devenir de la seconde extrémité de cette cassure (Fig. 4). Celle-ci peut en effet utiliser, pour son élongation, soit la première extrémité après extension, soit le deuxième brin de la région homologue.
Voie de synthèse dépendante de l’hybridation de brin
Après synthèse d’ADN au niveau de la première extrémité envahissante, le brin néo-synthétisé peut être déplacé de la boucle en D. Il peut alors s’hybrider avec la seconde extrémité de la cassure et être utilisé comme matrice pour allonger ce second brin, jusqu’à reconstitution d’un chromosome intact (Figure 4E). C’est la voie de synthèse dépendante de l’hybridation de brin, ou voie de déplacement de brin-hybridation de brin (SDSA).
Voie de réparation des cassures double brin
La seconde voie qui permet la résolution des intermédiaires de recombinaison est la voie dite de réparation des cassures double brin (DSBR). Le facteur de recombinaison Rad52 permet la capture d’une seconde extrémité d’ADN sur une boucle en D, ce qui forme une structure en double jonction de Holliday (dJH) (Fig. 4F). L’analogue fonctionnel de Rad52 chez l’Homme est BRCA2 dont les mutations sont impliquées dans de nombreux cancers du sein, ce qui souligne l’importance d’une bonne réparation de ces dommages à l’ADN.
Ces structures en double jonction de Holliday sont détectées in vivo au niveau d’une cassure induite dans les cellules en division mitotique, avec une préférence pour l’utilisation de la chromatide sœur comme région homologue, mais elles sont beaucoup moins fréquentes qu’en méiose. En effet, l’interaction entre le chromosome cassé et un chromosome homologue au niveau de la boucle en D peut résulter en un échange d’un fragment terminal entre les deux chromosomes, ce qui pourrait être délétère si les deux chromosomes n’étaient pas exactement identiques. Alors que ce phénomène d’échange, appelé « crossing-over », est fréquent en méiose où il permet le brassage génétique, il est rare lors d’une cassure en mitose. La faible fréquence des dJH en croissance végétative est donc compatible avec l’idée que les voies de recombinaison sans crossing-over sont favorisées dans les cellules en croissance végétative, contrairement à la méiose, puisque ce mécanisme crée un risque de crossing-over.
Ainsi, dans le DSBR, dans un premier temps, la seconde extrémité de la cassure est capturée par le second brin de la région homologue de la boucle en D. Puis elle est allongée par synthèse d’ADN avec la région homologue utilisée comme matrice (Fig. 4F). Cette structure en dJH peut alors être éliminée par deux mécanismes alternatifs, par résolution ou dissolution.
La résolution est réalisée par coupure simultanée des deux JH. La résolution peut résulter ou non en la formation de crossing-over, selon l’orientation de la coupure de chacune des jonctions de Holliday (Fig. 4F). Cette voie de résolution semble peu favorisée en dehors de la méiose, en raison du risque de crossing-over. Alternativement, un glissement convergent des deux JH permet leur dissolution sans crossing-over.
Ainsi, de nombreux mécanismes complexes, complémentaires les uns des autres et très régulés, permettent de réparer les CDBs avec une fidélité optimale.
La tolérance aux dommages
Une grande diversité de systèmes peut donc être mise en place afin de conserver l’intégrité du génome et restaurer fidèlement l’information génétique. Cependant, il arrive que les dommages à l’ADN aient lieu pendant la phase S, ou ne soient pas réparés avant la réplication, ce qui entraîne des problèmes lorsque la fourche de réplication arrive à leur niveau.
La réplication face aux dommages à l’ADN
La réplication semi-conservative est le mécanisme qui permet de dupliquer l’intégralité du génome (Branzei & Foiani, 2010 ; Watson et al, 2008b). Elle débute au niveau d’origines de réplication, régions dont la position est clairement définie chez S. cerevisiae mais pas dans d’autres organismes. Le complexe d’hélicases réplicatives progresse le long de l’ADN avec l’ensemble des protéines de réplication et ouvre la double hélice pour former une fourche de réplication bidirectionnelle associée au réplisome. La synthèse d’ADN est initiée par un court fragment d’ARN amorce, synthétisé puis allongé initialement en ADN par la Pol alpha-primase. Du fait de la chimie du processus, la synthèse de l’ADN est orientée de 5’ vers 3’. En conséquence, un brin d’ADN est synthétisé de manière continue depuis l’origine de réplication. L’autre brin, le brin discontinu, est répliqué dans le sens inverse de la progression de la fourche, sous forme de fragments d’Okazaki de quelques centaines de paires de bases. Des mutants de fidélité des différentes ADN polymérases ont montré que le brin continu est principalement synthétisé par la Pol epsilon, alors que la Pol delta réplique le brin discontinu. La processivité des ADN polymérases est favorisée par la présence du PCNA, une pince coulissante qui maintient l’association de ces polymérases à la fourche de réplication.
Cependant, de nombreux évènements peuvent créer une gêne pour la réplication, ce qui entraîne un ralentissement, voire un arrêt de la fourche. Par exemple, les zones de collision entre la fourche de réplication et la machinerie de transcription, ainsi que les régions répétées, capables de former des structures secondaires, sont des zones d’instabilité de la réplication. Lorsque les cellules sont soumises à des dommages UV, cela peut entraîner un découplage de la fourche de réplication, ce qui forme de longues régions simple brin. L’activation des points de contrôle cellulaires permet la stabilisation de ces fourches ralenties ou bloquées (Fig. 2).
La résolution de la réplication au niveau de ces zones difficiles peut se faire par plusieurs voies. Ainsi, chez S. pombe, la voie de recombinaison homologue permet de sauver les fourches de réplication bloquées au niveau d’une barrière de réplication, mais au risque de réarrangements chromosomiques. Certains dommages à l’ADN, tels ceux réparés par les voies de réparation par excision de base et par excision de nucléotides, créent des déformations des brins d’ADN ou des modifications de bases azotées qui ne peuvent pas être répliquées par les polymérases réplicatives, et risquent donc de bloquer la progression de la fourche de réplication, notamment au brin continu. La voie de tolérance aux dommages à l’ADN (DDT) permet à la fourche de réplication de dépasser ces erreurs sans les corriger.
Une réponse (in)fidèle : les polymérases translésionnelles
Lorsqu’une fourche de réplication rencontre une lésion simple brin sur l’ADN, il lui est donc nécessaire de trouver un moyen de la contourner. S’il lui est parfois possible de dépasser un dommage par réamorçage après la lésion, la synthèse translésionnelle, première modalité de la voie du DDT, propose une solution directement lors de la réplication. Ce processus est initié par une modification post-transcriptionnelle du PCNA (pince coulissante) par l’ubiquitine.
Mono-ubiquitinylation du PCNA
L’ubiquitine est une petite protéine régulatrice ubiquitaire de 8,5 kDa. Elle est utilisée pour modifier de manière post-transcriptionnelle de nombreuses protéines par formation de liaisons covalentes avec les lysines de ces protéines (voir le document sur l’ubiquitine). Sa fonction la plus connue permet le ciblage vers le protéasome, mais elle possède de nombreux autres rôles, selon le type de liaison créées avec la protéine cible. La réaction d’ubiquitinylation (ou ubiquitination) d’une protéine nécessite 3 étapes. La première étape consiste en une réaction d’activation de l’ubiquitine par une enzyme E1, avec consommation d’ATP. L’ubiquitine est alors liée de manière covalente à l’E1 par son extrémité C-terminale, avant d’être transférée sur l’enzyme de conjugaison d’ubiquitine E2. Une enzyme ubiquitine-ligase E3 permet alors d’associer l’E2 avec le substrat, ce qui permet le transfert de l’ubiquitine vers une lysine cible de la protéine substrat. Il existe plusieurs centaines d’E3, chacune spécifique d’un ou quelques substrats.
Le PCNA peut-être ubiquitinylé sur sa lysine 164, de préférence lorsqu’il est associé à l’ADN. Cette ubiquitinylation, qui a lieu spécifiquement en réponse aux dommages à l’ADN, est essentielle pour la voie de tolérance aux dommages (Fig. 5).
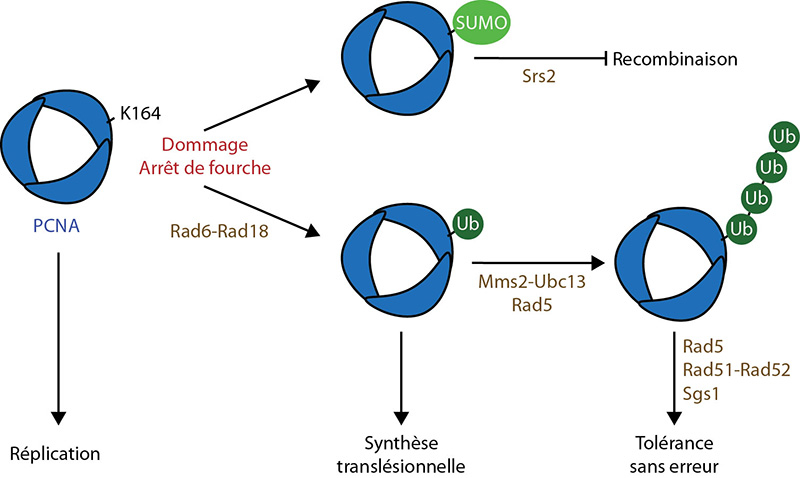
Lorsque la fourche de réplication rencontre un dommage, le PCNA est mono- puis poly-ubiquitinylé, sur sa lysine 164, ce qui entraîne différentes voies de tolérance aux dommages. La SUMOylation sur cette même lysine permet le recrutement de Srs2, qui empêche la recombinaison en désassemblant les filaments de Rad51.
Les principaux facteurs impliqués sont indiqués en marron. UB : Ubiquitine.
D'après : Boiteux & Jinks-Robertson, 2013 ; Ulrich, 2007 ; Zhang et al, 2011.
Synthèse translésionnelle
Cette mono-ubiquitinylation du PCNA permet le recrutement de polymérases dites translésionnelles qui, contrairement aux polymérases réplicatives, sont capables d’ajouter des nucléotides en face d’un dommage à l’ADN : Pol êta, Pol zêta et Rev1 (Fig. 6 A et B). Chacune de ces polymérases possède des dommages favoris, qu’elle est capable de répliquer sans faire d’erreurs. Ainsi, la Pol êta est capable d’ajouter correctement deux adénines face à un dimère de thymines résultant de l’action des UV, mais elle est peu fidèle et peu processive sur un substrat d’ADN normal. En conséquence, les mutants des gènes codant pour les protéines impliquées dans ce mécanisme et les polymérases translésionnelles présentent une diminution de la fréquence de mutation en réponse aux UV, de même qu’un mutant ponctuel du PCNA sur la lysine 164. Ceci montre que cette voie de synthèse translésionnelle, qui permet la complétion de la réplication en présence de dommages à l’ADN, crée des mutations, d’où sa dénomination comme voie de tolérance aux dommages source d’erreurs.
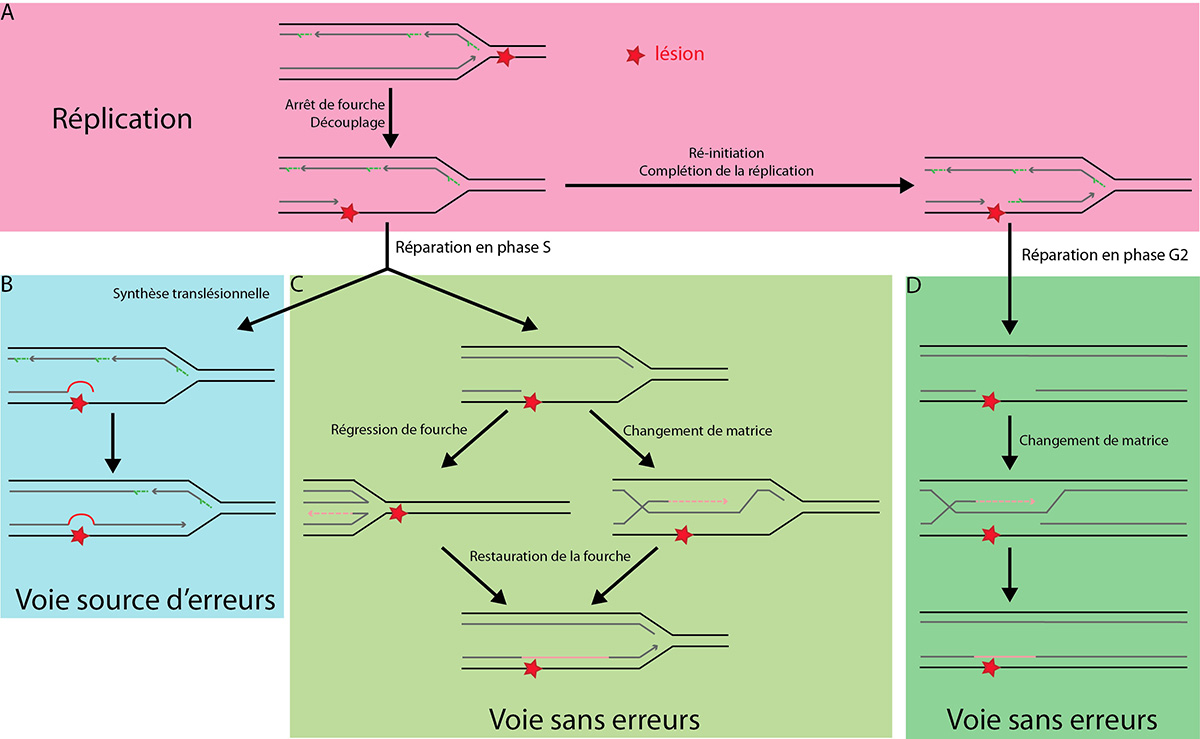
La rencontre d'un dommage sur le brin continu entraîne un arrêt de la fourche de réplication et un découplage entre brin continu et discontinu. Le dommage peut, soit être dépassé par réamorçage après celui-ci, pour être réparé après la réplication (A), soit la réplication est prolongée par des polymérases translésionnelles (B) ou par une voie sans erreurs (C). Un dommage au brin discontinu peut être ignoré par la réplication puis réparé en phase G2 par la voie sans erreurs, comme un dommage au brin continu après réamorçage (D).
D'après : Blastyak et al, 2007 ; Branzei & Foiani, 2010 ; KLein, 2007.
Le recrutement et l’activité des polymérases translésionnelles sont régulés, ce qui limite la fréquence des mutations. Les polymérases translésionnelles possèdent notamment des domaines d’interaction avec l’ubiquitine, nécessaire à leur activité translésionnelle. De plus, le PCNA peut favoriser leur activité. Ces polymérases peuvent aussi agir de manière découplée de la fourche de réplication, notamment en phase G2 et sur les régions simple brin laissées après le passage de la réplication. En phase G2, les levures mutées pour une des polymérases translésionnelles exposées aux UV forment des trous simple brin dans l’ADN, qui sont réparés par la recombinaison homologue. Ceci montre que les différentes voies de protection de l’ADN peuvent se complémenter.
La réponse sans erreur
Des gènes (UBC13, MMS2 et RAD5) ont aussi été identifiés dans la voie de tolérance aux dommages, notamment par leur sensibilité aux UV. Pourtant, contrairement aux polymérases translésionnelles, la perte de ces gènes conduit à l’augmentation de la fréquence de mutations, ce qui suggère qu’ils sont impliqués dans une voie sans erreur, qui constitue la seconde modalité du DDT. De plus, ces gènes sont, avec d’autres, importants pour protéger la réplication en cas de stress et en réponse aux UV, ce qui montre une voie essentielle dans la complétion de la réplication.
Poly-ubiquitinylation du PCNA
Une fois mono-ubiquitinylé en réponse à un dommage, le PCNA peut être poly-ubiquitinylé par formation de chaînes K63 de l’ubiquitine sur sa lysine 164 (Fig. 5). Cette poly-ubiquitinylation est nécessaire pour la fonction du PCNA dans la voie de tolérance aux dommages sans erreur.
Réparation par changement de matrice
La poly-ubiquitinylation du PCNA permet la mise en place de cette voie de réparation sans erreur, par un mécanisme qui semble utiliser la chromatide-sœur comme matrice pour la synthèse d’ADN, ce qui entraîne donc la copie à l’identique de la séquence face à la lésion. Différents modèles sont actuellement proposés pour ce mécanisme (Fig. 6).
Un premier modèle propose une réversion de fourche au niveau du brin continu bloqué par un dommage. Dans ce modèle, le brin néo-synthétisé de la chromatide-sœur, allongé par découplage de la fourche de réplication, est hybridé avec le brin naissant bloqué au niveau de la lésion par un phénomène de recul de la machinerie de réplication. Il peut alors être utilisé comme matrice pour la synthèse au-delà du dommage (Fig. 6C).
Un second modèle possible pour la réparation sans erreur serait la formation de jonctions entre chromatides-sœurs, par un mécanisme d’invasion de la chromatide-sœur. Comme lors de la recombinaison homologue, cette structure permet alors d’utiliser la chromatide-sœur comme matrice pour allonger l’ADN par la Pol delta et ainsi dépasser le dommage en maintenant la continuité du brin d’ADN, avant d’être résolue (Fig. 6C).
Rad5 est la principale protéine identifiée dans cette voie sans erreur. In vitro, Rad5 est ainsi capable d’inverser et de faire régresser des modèles de fourche de réplication, mais aussi de permettre l’invasion de brin.
Rad5 est nécessaire au redémarrage des fourches bloquées lorsque les cellules sont exposées à l’adozelesin, un agent alkylant de l’ADN. Les deux activités de Rad5, E3 ligase et ATPase, sont importantes pour cette protection, qui passe par la formation de structures en X dépendantes de Rad5 et contenant des jonctions de Holliday. Ces structures en X, qui peuvent correspondre à l’invasion de la chromatide-sœur par le brin à allonger, dépendent aussi de l’activité de la polymérase réplicative Pol delta, mais pas des polymérases translésionnelles. Ainsi, la formation de structures particulières, qui implique à la fois des facteurs de la voie de tolérance et de la recombinaison, est nécessaire pour finaliser la réplication en réponse à certaines lésions à l’ADN.
Une dissolution sans erreur de ces structures intermédiaires de l’ADN est favorisée pendant la réplication, permettant de résoudre les structures restantes avant la division cellulaire, mais en favorisant des crossing-over au risque de réarrangements chromosomiques.
Lorsque certaines des protéines de la voie de tolérance aux dommages, comme Rad5, sont restreintes expérimentalement en phase G2 du cycle cellulaire, cette voie est toujours fonctionnelle, et cela n’empêche pas la complétion globale de réplication. De plus, un dommage au brin discontinu ne bloque pas la fourche de réplication, du fait même de la nature discontinue de cette synthèse, et il a été montré qu’une fourche de réplication peut ré-initier la synthèse au brin continu après un dommage. Les facteurs de recombinaison favorisent la résolution des régions simple brin laissées par les fourches de réplication, qui peuvent correspondre à des sites de lésions non réplicables. En outre, le chargement sur l’ADN de Rad51 et Rad52 au niveau de la fourche de réplication, au cours de la phase S, est nécessaire à la tolérance aux dommages via ces protéines, même si la réparation en elle-même est faite plus tardivement, en phase G2.
La fourche de réplication peut aussi s’adapter et dépasser certains dommages à l’ADN pour terminer la copie du génome avant de mettre en place le mécanisme de tolérance. Néanmoins, cette ignorance du dommage laisse une région simple brin non répliquée, ce qui peut expliquer pourquoi cette voie, aussi appelée voie de réparation post-réplication (PRR), peut agir après la réplication, par un mécanisme éventuellement indépendant de la fourche de réplication. Ce mécanisme de tolérance, qui peut impliquer à la fois les facteurs du DDT et de la HR, pourrait se faire par un phénomène de changement de matrice avec la chromatide-sœur similaire à celui proposé en phase S (Fig. 6D).
En conclusion, la voie de tolérance aux dommages est une voie qui permet la réplication complète du génome, en synthétisant une séquence d’ADN la plus fidèle possible face à une lésion à l’ADN. Ce mécanisme rétablit de surcroît la continuité du brin d’ADN néo-synthétisé, ce qui est nécessaire à la transmission d’une information génétique exacte à la descendance et pour la correction des lésions après la réplication.
Conclusion
Les dommages à l’ADN font ainsi partie de la vie d’une cellule et peuvent être à l’origine de nombreux problèmes délétères, d’où l’importance pour celle-ci d’avoir des mécanismes de réparation puissants, spécialisés et efficaces. Lorsque des mutations ponctuelles non réparées surviennent dans les lignées germinales, elles sont alors transmises à la descendance et sont à l’origine des maladies génétiques, parfois graves. La dérégulation des mécanismes de réparation et de signalisation est la source de l’instabilité génétique observée dans les cancers, et participe ainsi à la carcinogénèse. Pourtant, toutes les mutations ne sont pas délétères : certaines n’ont pas d’effet, et d’autres sont la source de la variabilité génétique entre individus. Certaines de ces mutations peuvent même être plus favorables à la survie de l’individu dans son environnement donné, ce qui favorisera alors la transmission de la mutation favorable à sa descendance par la sélection naturelle. Il y a ainsi une concurrence permanente entre le risque encouru par l’individu et le gain potentiel pour l’espèce. Les dommages à l’ADN réparés avec des erreurs sont donc un moteur essentiel à l’évolution des espèces.
Références
Remerciements à Maria Teresa Teixeira, responsable du groupe Biologie des Télomères au laboratoire de Biologie Moléculaire et Cellulaire des Eucaryotes (UMR8226 CNRS/UPMC).
Références citées dans le texte :
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3 : 958-965.
- Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L (2007) Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell 28 : 167-175.
- Boiteux S, Jinks-Robertson S (2013) DNA Repair Mechanisms and the Bypass of DNA Damage in Saccharomyces cerevisiae. Genetics 193 : 1025-1064.
- Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11 : 208-219.
- Dulbecco R (1949) Reactivation of ultra-violet-inactivated bacteriophage by visible light. Nature 163 : 949.
- Finn K, Lowndes NF, Grenon M (2012) Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci 69 : 1447-1473.
- Franklin RE, Gosling RG (1953) Molecular configuration in sodium thymonucleate. Nature 171 : 740-741.
- Harrison JC, Haber JE (2006) Surviving the breakup : the DNA damage checkpoint. Annu Rev Genet 40 : 209-235.
- Houtgraaf JH, Versmissen J, van der Giessen WJ (2006) A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med 7 : 165-172.
- Kelner A (1949) Effect of Visible Light on the Recovery of Streptomyces Griseus Conidia from Ultra-violet Irradiation Injury. Proc Natl Acad Sci USA 35 : 73-79.
- Klein HL (2007) Reversal of fortune : Rad5 to the rescue. Mol Cell 28 : 181-183.
- Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38 : 233-271.
- San Filippo J, Sung P, Klein H (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 : 229-257.
- Symington LS, Gautier J (2011) Double-strand break end resection and repair pathway choice. Annu Rev Genet 45 : 247-271.
- Ulrich HD (2007) Conservation of DNA damage tolerance pathways from yeast to humans. Biochem Soc Trans 35 : 1334-1337.
- Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2008b) The replication of DNA. In Molecular Biology of the Gene – Sixth Edition, Edition PI (ed).
- Watson JD, Crick FH (1953a) Genetical implications of the structure of deoxyribonucleic acid. Nature 171 : 964-967.
- Watson JD, Crick FH (1953b) Molecular structure of nucleic acids ; a structure for deoxyribose nucleic acid. Nature 171 : 737-738.
- Wilkins MH, Stokes AR, Wilson HR (1953) Molecular structure of deoxypentose nucleic acids. Nature 171 : 738-740.
- Zhang W, Qin Z, Zhang X, Xiao W (2011) Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS Lett 585 : 2786-2794.
Pour aller plus loin
The DNA Damage Response : Making It Safe to Play with Knives, Alberto Ciccia et Stephen J. Elledge
Article de 2010 qui fournit notamment des informations sur différentes sources de dommages à l’ADN provenant de l’environnement (dommages causés par la cigarette, un vol en avion, une exposition à Tchernobyl…)