Qu'est-ce qu'un prion ? Quelle est sa structure et comment celle-ci participe-t-elle à sa pathogénicité ?
Le prion (acronyme de proteinaceous infectious particles) (Prusiner, 1982) est l’agent responsable des encéphalopathies spongiformes transmissibles (EST) ou maladies à prion. Ces maladies neurodégénératives touchent la plupart des mammifères notamment les bovins (ESB pour encéphalopathie spongiforme bovine ou maladie de la vache folle), les petits ruminants (tremblante), certains cervidés sauvages (maladie du dépérissement chronique) et l’Homme (kuru, maladie de Creutzfeldt-Jakob (MCJ)).
Au plan neuropathologique, ces maladies se caractérisent par une mort neuronale conférant l’aspect spongieux du cerveau, accompagnée d’une prolifération anormale des cellules gliales du système nerveux central, appelée gliose, (Figure1) ainsi que d’un dépôt protéique fibrillaire extracellulaire (ou plaques amyloïdes, appelées ainsi car elles ressemblent à des grains d’amidon) composé quasi-exclusivement de la protéine PrP du prion (ou PrPSc, voir plus bas).
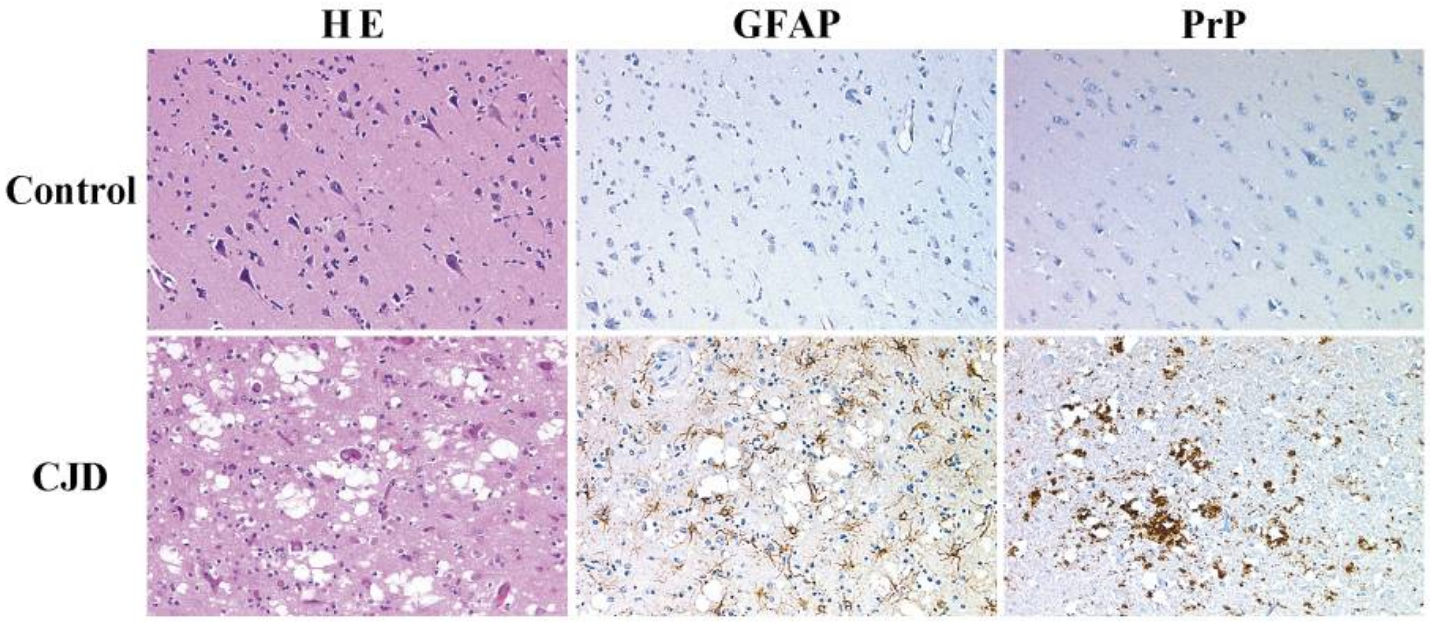
Coupes histologiques et immunohistologiques de cortex frontal provenant d’un patient décédé sans syndrome neurologique (control), ou d’un patient atteint de la maladie de Creutzfeldt-Jakob (CJD). Les sections de cerveaux sont colorées avec l’hématoxyline-éosine (HE, colonne de gauche – l’hématoxyline colore les noyaux en bleu-violet, l’éosine révèle le cytoplasme en rose), ou marquées avec un anticorps reconnaissant la protéine GFAP des cellules gliales (colonne du milieu), ou marquées avec un anticorps reconnaissant la protéine PrP (colonne de droite). Chez le patient malade, la perte neuronale et la spongiose sont visibles à la coloration HE (taches blanches). Une forte prolifération des cellules gliales (nommée gliose) et des dépôts de PrP sont détectés respectivement par le marquage des anticorps anti-GFAP et anti-PrP des sections de cortex de patient atteints de MCJ.
Le prion comme agent pathogène… tout un concept !
Le caractère transmissible de ces maladies fut démontré en 1936. Deux vétérinaires français, Cuillé et Chelle transmirent avec succès la tremblante du mouton à un animal sain par inoculation intraoculaire d’un homogénat de moelle épinière provenant d’un animal malade. Mais la nature de l’agent infectieux fut longtemps débattue. Qualifié dans un premier temps « d’agent non conventionnel » parce que ne répondant pas aux méthodes de caractérisation des agents pathogènes classiques (virus ou bactérie), c’est en 1967 que le mathématicien Griffith émit pour la première fois l’hypothèse que cet agent pourrait être uniquement constitué d’une protéine. Celle-ci existerait sous deux formes : une forme pathologique (anormale) qui pourrait transmettre son information de structure à la forme non-pathologique (normale) et catalyser ainsi le changement de conformation vers la forme pathogène (Griffith, 1967). 40 ans plus tard, Stanley Prusiner démontra la nature exclusivement protéique de cet agent infectieux, constitué uniquement de la protéine PrP anormale, celle-là même que l’on retrouve au niveau des dépôts cérébraux (Prusiner, 1993).
Il fut alors démontré que les maladies à prions étaient causées par le mauvais repliement d’une protéine de l’hôte : la protéine PrP. Dans son état physiologique ou natif (noté PrPC pour « PrP cellulaire »), cette protéine est monomérique et ancrée à la face externe de la membrane plasmique de la cellule. La PrPC est impliquée dans de nombreuses fonctions biologiques, notamment dans la transduction de signaux cellulaires, l’homéostasie du cuivre ou le stress oxydatif. Dans sa conformation native, cette protéine est constituée de deux domaines, l’un non structuré, l’autre, globulaire formé de 3 hélices alpha. En revanche, au cours de la maladie, si la séquence primaire de la protéine est conservée, les structures secondaires, tertiaires et quaternaires (ces dernières correspondant au repliement spatial de la chaîne d’acides aminés ainsi que leurs assemblages de plusieurs sous-unités) se retrouvent complètement bouleversées. Le domaine globulaire perd ses hélices alpha et forme des feuillets bêta (Figure 2). Ce changement conformationnel est lourd de conséquences d’un point de vue physico-chimique. Alors que la protéine dans son état natif est soluble et sensible aux protéases, elle devient insoluble et résistante aux protéases. Mais surtout, les feuillets β confèrent à la protéine une forte propension à s’agréger et à former des assemblages fibrillaires amyloïdes comme ceux observés dans les tissus atteints. Le point le plus remarquable, est que la forme pathologique (notée PrPSc pour « PrP scrapie », terme anglais désignant la tremblante du mouton), à elle seule, est capable de forcer le mauvais repliement de la forme native vers la forme pathogène. On dit que ce processus de repliement est auto-réplicatif et c’est ce caractère qui a donné naissance au concept de prion.
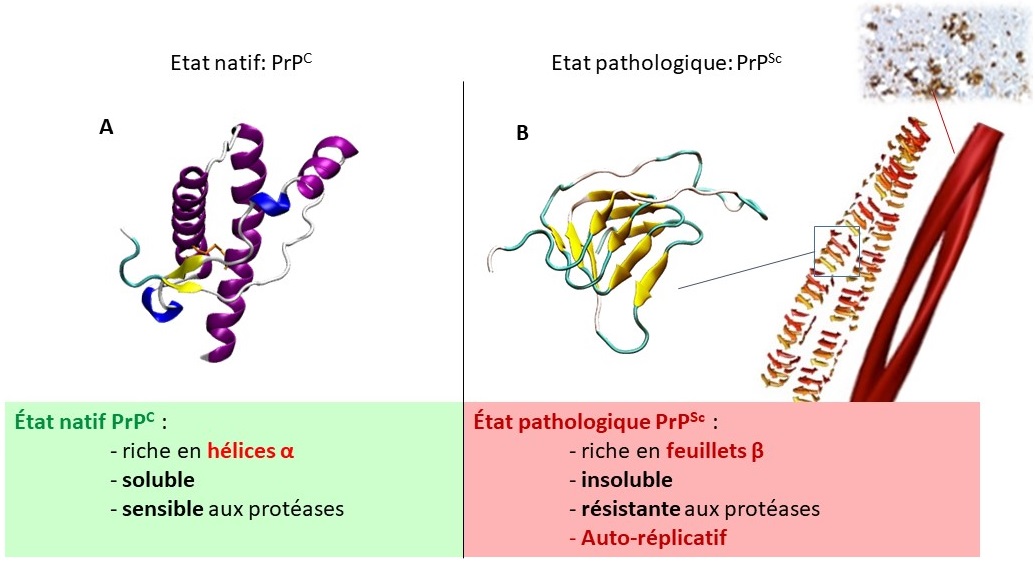
A. Structure tertiaire du domaine globulaire de la PrPC (résidus ̴125-230). Les hélices α sont représentées en violet et les feuillets antiparallèles en jaune. Licence : CC-BY-NC
B. Modèle de la structure tertiaire de la protéine PrPSc (à gauche) qui présente un tonneau β. En s’agrégeant, les protéines PrPSc donnent naissance à des structures quaternaires (à droite) : les fibres amyloïdes. Licence : CC-BY
Plus surprenant encore, au sein de la même espèce hôte, les maladies à prion peuvent donner lieu à tout un panel de phénotypes. Ces variations physiopathologiques incluent le temps d’incubation de la maladie (qui peut être extrêmement long), le profil lésionnel (tropisme tissulaire, distribution des dépôts de PrPSc et de la vacuolisation) ainsi que leurs propriétés biochimiques (profil de glycosylation, degré de résistance à la protéolyse et à la dénaturation, site de coupure des protéases…). Ces caractères sont stables et conservés aux cours des transmissions successives. Ils permettent de définir ce qui est communément appelé le phénomène de souche, par analogie avec les agents infectieux conventionnels. Or, comment expliquer qu’une particule infectieuse, dépourvue de génome et composée d’une seule et même protéine, puisse induire des variabilités phénotypiques si marquées ? L’explication la plus plausible est que ces particules puissent arborer tout une panoplie de conformations. L’information structurale constitue donc l’information pathogène où un type (ou un mélange de sous-types) de conformation engendrerait un phénotype de maladie.
Le prion comme agent zoonotique… une question de compatibilité structurale
Les prions ne sont pas seulement capables de se propager d’une cellule à une autre, mais également entre individus, et ce, qu’ils soient de la même espèce ou non. L’exemple le plus tristement célèbre est celui de l’encéphalopathie spongiforme bovine (ESB). Largement médiatisée, la transmission de l’agent prion responsable de l’ESB fut à l’origine d’une crise économique et sanitaire majeure entre les années 1980 et 2000. Qualifiée de « crise de la vache folle » à cause du tableau clinique des bovins atteints qui inclue un comportement agressif, une hyperréactivité aux stimuli et des troubles moteurs (tremblement, déséquilibre, trouble de la démarche), elle sévit majoritairement en Europe. Les premiers cas furent diagnostiqués au Royaume-Uni en 1986 (Wells et al., 1987). Sa progression fut fulgurante dans ce pays, passant de 446 nouveaux cas recensés en 1987 à 37 000 pour la seule année 1993. Comment le prion a-t-il pu se propager si vite ? Il faudra attendre deux ans après l’apparition des premiers cas pour que des épidémiologistes britanniques cernent la cause de l’épidémie : une exposition du cheptel bovin à un agent prion présent dans les farines animales (FVO ; farine de viande et d’os) destinées à l’alimentation des bovins (Wilesmith et al., 1988). À cette époque, les cadavres de ruminants trouvés en exploitations ainsi que les produits d’équarrissage étaient utilisés pour la fabrication de ces farines, ce qui constituait une source de protéines bon marché. Afin d’augmenter leurs marges bénéficiaires, les barèmes de pressions/températures destinés à inactiver tout agent pathogène dans ces farines ont progressivement été baissés. Le protocole de fabrication ne permettant plus l’éradication du prion, l’agent a donc été distribué à grande échelle par le biais de recyclage des carcasses infectées, contaminant ainsi un nombre croissant d’animaux. À titre indicatif, un cerveau de souris contaminée contient suffisamment d’infectiosité pour transmettre la maladie à un million de congénères. Au total, plus de 184 624 cas d’ESB ont été confirmés au Royaume-Uni [1]. Le reste de l’Europe, ne fut pas épargné : en France c’est 274 cas d’ESB qui furent recensés en 2001.
Parallèlement, en 1996, les médecins britanniques rapportent 10 cas d’une nouvelle forme de la maladie de Creutzfeldt-Jakob chez l’Homme (Will et al., 1996). Appelée variant de la maladie de Creutzfeldt-Jakob (vMCJ), le tableau clinique observé était clairement différent des formes classiques de la MCJ dite sporadique, mais très homogène parmi ces 10 nouveaux cas. Ceci permettait de suspecter une origine commune du vMCJ. L’élément le plus inquiétant concernait l’âge des malades : la majorité avait moins de 30 ans contre 65 pour les formes classiques. Le fait de recenser une dizaine de cas en quelques mois paraissait hautement suspect dans un pays frappé depuis dix ans par la maladie de la vache folle. Dans les mois qui suivirent, des résultats expérimentaux permirent d’établir clairement le lien avec l’épizootie d’ESB suite à l’inoculation expérimentale à des macaques de l’agent de l’ESB qui entraîna chez ces animaux une EST présentant de nombreux points communs avec la vMCJ : lésions similaires (présence de plaques amyloïdes (Lasmézas et al., 1996) et signature biochimique de la PrPSc en gel de polyacrylamide spécifique (Collinge, et al. 1996). On estime à 180 le nombre de patient atteints de vMCJ au Royaume-Uni. La crise de la vache folle fut la démonstration que les prions pouvaient se transmettre naturellement des bovins à l’Homme via la chaîne alimentaire, requalifiant les prions d’agents zoonotiques (une zoonose est une maladie infectieuse transmissible des animaux aux êtres humains, et réciproquement). 27 cas furent recensés en France.
Heureusement, la contamination inter-espèce n’est pas aussi facile pour tous les prions et pour toutes les espèces. En règle générale, il existe une barrière de transmission de la particule infectieuse prion entre les espèces, celles-ci étant plus ou moins permissives aux différents prions selon la séquence primaire de la PrPC de l’hôte. Ainsi des espèces comme le lapin, le chien, le porc et le cheval, semblent être résistantes à la plupart des prions issus d’espèces différentes, contrairement aux hamsters ou aux campagnols qui sont très permissifs. La barrière d’espèce semble donc être largement due aux différences de conformation imposées par la séquence primaire de la protéine. C’est ce qu’on appelle, la barrière de séquence. La propagation du prion nécessite le recrutement et la conversion de la PrPC de l’hôte en PrPSc. Le degré de similitude de la séquence primaire entre la PrPSc inoculée et la PrPC de l’hôte est un élément clé de l’efficacité de transmission inter-espèce (Prusiner et al., 1990). Cette hypothèse fut validée par les travaux de Scott (Scott et al., 1993). Il montra qu’il était en mesure d’infecter des souris (normalement non-permissives à la souche de prion PrPSc de hamster) en construisant des souris transgéniques exprimant la PrPC du hamster. Il y a donc généralement absence de barrière d’espèce pendant une transmission homotypique c’est-à-dire lorsque les séquences des protéines PrPC des deux espèces sont proches.
La structure des prions… la quête du Saint Graal pour les chercheurs
Conceptuellement, l’adaptation du prion à son nouvel hôte au cours d’une transmission s’appuierait sur l’ajustement structural entre la PrPSc infectante et la PrPC de l’hôte. Ainsi, la détermination à l’échelle atomique de la structure de la PrPSc se trouverait être la clé pour comprendre le fonctionnement du prion et permettrait de répondre aux questions toujours d’actualité telles que : à quel niveau structural l’information de souche est-elle codée ? Comment cette information se perpétue-t-elle ?
Le problème est que les propriétés physico-chimiques des prions rendent cette tâche pour le moment difficile. En effet, la cristallographie aux rayons X nécessite un échantillon très homogène, cristallin, pour déterminer précisément la structure d’une protéine, ce qui est loin d’être possible pour les prions. D’autres techniques indirectes comme la cryo-microscopie recoupée avec des données de RMN (résonance magnétique nucléaire) suggèrent une structuration de la protéine PrPSc en tonneau β (figure 2B).
Il apparaît à présent que les fibres de prions présentent une organisation complexe avec l’existence d’une sous-unité élémentaire constituée de plusieurs PrPSc (de l’ordre du trimère) (Figure 3) (Igel-Egalon et al., 2017). Cette sous-unité est particulièrement stable : même après un traitement dénaturant (urée en concentration > 6 mol.L-1), elle conserve la capacité de se réassembler en PrPSc et surtout conserve toutes les informations de la souche de prion de départ. Par ailleurs, nous avons également démontré que cette brique élémentaire existe en équilibre naturel avec les fibres de PrPSc. Cette observation remet en cause l’idée que les fibres amyloïdes prion correspondent à des assemblages stables et pérennes où la PrPSc serait piégée. Au contraire, les fibres seraient des entités dynamiques qui relargueraient des unités élémentaires ultra-stables possédant les informations nécessaires à la propagation de l’information de souche. Ces unités pourraient-elles alors être le vecteur de diffusion au sein de l’organisme via des simples mouvements browniens ? L’existence de cette organisation soulève d’autres questions notamment celle de l’intégration des monomères de PrPC dans ces assemblages complexes : s’intègrent-ils de manière concerté dans l’assemblage ou bien un à un (figure 3c) ?
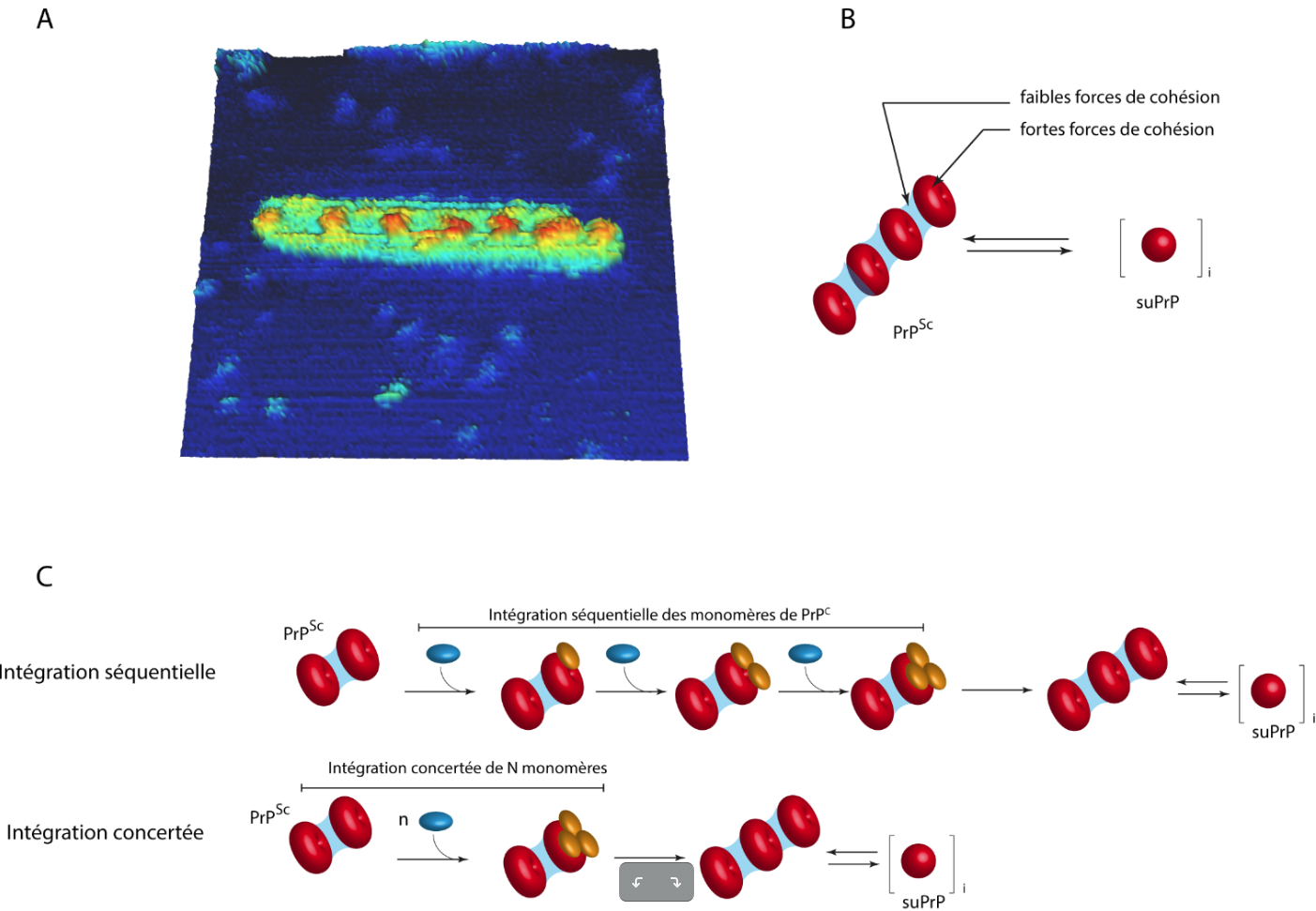
A. Cliché en microscopie à force atomique (AFM) d’une fibre recombinante de PrP humaine révélant l’existence d’un motif répété correspondant à l’unité élémentaire suPrP.
B. L’unité élémentaire suPrP (donut rouge) est constituée d’au moins 3 molécules de PrP. La cohésion des assemblages, en fibres de PrPSc (à gauche) ou en suPrP (à droite) fait intervenir différents types de forces qui expliquent l’existence d’un équilibre entre les deux états.
C. L’intégration des monomères de PrPC (bleu) reste à ce jour un phénomène non-connu. Deux hypothèses sont proposées : (ligne du haut) une intégration séquentielle des monomères au niveau d’une interface de polymérisation de la fibre, impliquant un changement de conformation de la forme normale PrPC (bleu) vers la forme anormale PrPSc (jaune) ; (ligne du bas) une intégration concertée de 3 monomères à la fois.
Pour conclure, 40 ans après l’émergence du concept de prion et l’idée qu’une protéine à elle seule puisse se transmettre, muter, et perdurer, cet agent pathogène reste toujours une énigme pour la Science. Mais percer les clés de son mystère n’a jamais été autant d’actualité notamment suite aux récents travaux menés sur d’autres protéinopathies comme les maladies d’Alzheimer et de Parkinson qui semblent partager des propriétés avec les prions sur bien des aspects. Les prions apparaissent être un modèle clef pour l’étude des maladies neurodégénératives amyloïdes au sens large.
Pour en savoir plus
- Corinne Ida Lazmesas : Qu’est-ce qu’un prion ; Le Pommier, 2005
- Stanley Prusiner : La Mémoire et la Folie. La découverte des Prions, un nouveau paradigme biologique. Odile Jacob, 2015
- Mohammed Moudjou : Le Mouton, La Vache et le vieux Papou, l’histoire d’une mauvaise graine. Edilivre, 2014
- Angélique Igel-Egalon et al. : Risques microbiologiques alimentaires, Chapitre 30 Prion : un agent infectieux de nature protéique. Lavoisier, 2017
Références
- Aguzzi, A. (2001). Recent developments in the pathogenesis, diagnosis, and therapy of prion diseases. Dialogues in Clinical Neuroscience, 3(1), 25–36. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/22034459
- Aguzzi, A., Sigurdson, C., & Heikenwaelder, M. (2008). Molecular mechanisms of prion pathogenesis. Annual Review of Pathology, 3(1), 11–40. https://doi.org/10.1146/annurev.pathmechdis.3.121806.154326
- Collinge, J., Sidle, K. C., Meads, J., Ironside, J., & Hill, A. F. (1996). Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature, 383(6602), 685–90. https://doi.org/10.1038/383685a0
- Govaerts, C., Wille, H., Prusiner, S. B., & Cohen, F. E. (2004). Evidence for assembly of prions with left-handed -helices into trimers. Proceedings of the National Academy of Sciences, 101(22), 8342–8347. https://doi.org/10.1073/pnas.0402254101
- Griffith, J. S. (1967). Self-replication and scrapie. Nature, 215(5105), 1043–4. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/4964084
- Igel-Egalon, A., Moudjou, M., Martin, D., Busley, A., Knäpple, T., Herzog, L., … Rezaei, H. (2017). Reversible unfolding of infectious prion assemblies reveals the existence of an oligomeric elementary brick. PLOS Pathogens, 13(9), e1006557. https://doi.org/10.1371/journal.ppat.1006557
- Lasmézas, C. I., Deslys, J.-P., Demaimay, R., Adjou, K. T., Lamoury, F., Dormont, D., … Hauw, J.-J. (1996). BSE transmission to macaques. Nature, 381(6585), 743–744. https://doi.org/10.1038/381743a0
- Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science (New York, N.Y.), 216(4542), 136–44. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/6801762
- Prusiner, S. B. (1993). Transgenetic investigations of prion diseases of humans and animals. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 339(1288), 239–54. https://doi.org/10.1098/rstb.1993.0022
- Prusiner, S. B., Scott, M., Foster, D., Pan, K. M., Groth, D., Mirenda, C., … Carlson, G. A. (1990). Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell, 63(4), 673–86. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/1977523
- Scott, M., Groth, D., Foster, D., Torchia, M., Yang, S. L., DeArmond, S. J., & Prusiner, S. B. (1993). Propagation of prions with artificial properties in transgenic mice expressing chimeric PrP genes. Cell, 73(5), 979–88. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8098995
- Wells, G. A., Scott, A. C., Johnson, C. T., Gunning, R. F., Hancock, R. D., Jeffrey, M., … Bradley, R. (1987). A novel progressive spongiform encephalopathy in cattle. The Veterinary Record, 121(18), 419–20. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3424605
- Wilesmith, J. W., Wells, G. A., Cranwell, M. P., & Ryan, J. B. (1988). Bovine spongiform encephalopathy: epidemiological studies. The Veterinary Record, 123(25), 638–44. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3218047
- Will, R. G., Ironside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K., Alperovitch, A., … Smith, P. G. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet (London, England), 347(9006), 921–5. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8598754
- Zweckstetter, M., Requena, J. R., & Wille, H. (2017). Elucidating the structure of an infectious protein. PLoS Pathogens, 13(4), e1006229. https://doi.org/10.1371/journal.ppat.1006229